Syllabus (Fourth Edition, 2023)
Topics
i. Describe the pharmacology of drugs used to treat pain.
ii. Outline the classification, structure and distribution of opioid receptors and NMDA receptors.
Topics not covered in previous SAQs
i. Describe the pharmacology of drugs used to treat pain. (not all drugs in pharmacopeia asked before)
ii. Outline the classification, structure and distribution of opioid receptors and NMDA receptors.
Learning Objectives for the First Part Examination in Intensive Care Medicine
- This will ensure that trainees, tutors, and examiners can work from a common base.
- All examination questions are based around this Syllabus.
- These learning objectives are designed to outline the minimum level of understanding required for each topic.
- The accompanying texts are recommended on the basis that the material contained within them provides sufficient information for trainees to meet the learning objectives.
- Trainees are strongly encouraged to explore the existing and evolving body of knowledge of the Basic Sciences as they apply to Intensive Care Medicine by reading widely.
- For all sections of the syllabus an understanding of normal physiology and physiology at extremes of age, obesity, pregnancy (including foetal) and disease (particularly critical illness) is expected.
- Similarly, for pharmacology, trainees are expected to understand a drug’s pharmacology in these contexts.
- An understanding of potential toxicity and relevant antidotes is also expected.
Definitions
Throughout the document specific wording has been used under the required abilities to indicate the level of knowledge and understanding expected and a glossary of these terms is provided.
Definitions
| Calculate | Work out or estimate using mathematical principles. |
| Classify | Divide into categories; organise, arrange. |
| Compare and contrast | Examine similarities and differences. |
| Define | Give the precise meaning. |
| Describe | Give a detailed account of. |
| Explain | Make plain. |
| Interpret | Explain the meaning or significance. |
| Outline | Provide a summary of the important points. |
| Relate | Show a connection between. |
| Understand | Appreciate the details of; comprehend. |
SAQs
i. Describe the pharmacology of drugs used to treat pain.
Opiates
2014A 24 – 2010A 24
Describe the mechanism of action of the analgesic effect of opiates. (70% of marks) Explain the mechanism by which morphine causes respiratory depression and constipation. (30% of marks)
CICMWrecks Answer
Mechanism of Action and Effects
- Opiates act on MOP, DOP, KOP and NOP receptors
- MOP are distributed widely:
- Cortex
- Basal ganglia
- Spinal cord – dorsal horn
- Periaqueductal grey – descending inhibition
- DOP, KOP and NOP less affected by commonly used opiates
- Produce analgesia, with different levels of other opiate effects
- Opioid receptors are GiPCR
- Inhibit Ca2+ channels
- Prevent neurotransmitter release at pre-synaptic nerve terminals
- Open K+ channels
- Hyperpolarise post-synaptic nerves, preventing signal transmission
- Inhibit Ca2+ channels
Analgesia
- Analgesia induced by inhibition of ascending pain transmission
- Sites of action
- Primary afferents
- Dorsal horn interneurons
- Descending modulatory neurons
- Inhibition of GABA-releasing inhibitory neurons
- Supraspinally
- Analgesia enhanced by centrally mediated euphoria and sedation
Respiratory depression
- MOP-mediated
- Action on medullary chemoreceptors
- Blunted increase in respiratory drive with increasing CO2
- Effect is antagonised by painful stimulus
Constipation
- Mediated by MOP, KOP and DOP receptors
- Expressed in high density throughout GIT nerve plexus
- Increased GIT tone, with subsequent loss of rhythmic peristaltic movements
Mooney 2016
Examiner Comments
2014A 24: 11% of candidates passed this question.
This question was poorly answered with a low pass rate; which was unexpected for this core topic. In general, completed answers demonstrated good understanding of the analgesic mechanisms of opiates, but only superficial understanding of respiratory depression and constipation. Better answers explained the decreased responsiveness of the respiratory centre to CO2, and the effects of opiates on the enteric nervous system and peristalsis.
2010A 24: 3 (30%) of candidates passed this question
This was a multi-part question, for which many candidates failed to apportion their time as indicated by the question. Most patients who enter Intensive Care receive opioid analgesia so candidates were expected to have detailed knowledge about the mechanics of action of opiates. For a good answer candidates were expected to mention that opiate agonists produce analgesia by binding to Mu receptors (which are G protein coupled) in the central and peripheral nervous system and spinal cord and their cellular mechanism of action eg presynaptic neurone – close voltage gated Ca channels and prevent neurotransmitter release and post synaptic neurone – hyperpolarise and inhibit post synaptic neurone. The fact that opiates also affect emotional side of pain and cause euphoria which may help with pain perception was often omitted.
Mechanism of respiratory depression is mediated via mu receptor. It occurs at normal analgesic doses, and decreases the chemosensitivity of the respiratory centre to PaCO2
Mechanism of constipation results from increased tone and decreased motility of the GIT via action on visceral smooth muscle mediated by intramural nerve plexus and all three opioid receptors.
Many candidates had poor organisation and poor knowledge of all aspects of this question.
Syllabus: G2d 2b
Reference: Pharmacology, Rang and Dale pgs 600-601, Basic and Clinical Pharmacology, Katzung pg 492
2018A 05
Compare and contrast the pharmacokinetics and pharmacodynamics of IV fentanyl and IV remifentanil (60% of marks). Discuss the concept of context sensitive half-time using these drugs as examples (40% of marks).
CICMWrecks Answer
PK and PD of Fentanyl and Remifentanil
Context sensitive half-time
- Defined as the time for plasma concentration to fall to half of its value at the time of stopping an infusion
- A method to describe the variability in plasma concentrations after ceasing an infusion
The “context” is the duration of infusion. - Used because terminal elimination half-life has little clinical utility for predicting drug offset
Half-lives are often misleading when discussing drug infusions. - Dependent on:
- Duration of infusion
During an infusion, drugs distribute out of plasma into tissues. When the infusion ceases, drug is cleared from plasma and tissue drug redistributes back into plasma.- The longer an infusion, the more drug has distributed out of tissues, and the longer the redistribution phase
- The longest context-sensitive half time occurs when an infusion is at steady-state
- Redistribution
The maximal CSHT reached depends on the:- VDss
Drugs with a larger VDss have a longer CSHT, as only a small proportion of the drug in the body will be in plasma and able to be cleared. - Rate constant for elimination
Drugs with a smaller rate constant for elimination have a longer CSHT.
- VDss
- Duration of infusion
Drugs with longer context-sensitive half-times will wear off less predictably.
Context sensitive half time – Fentanyl vs Remifentanil:
- Fentanyl shows variable CSHT – Has high redistribution, with VdSS 3-5L/kg, and a terminal half life of 3 hours. Hence, prolonged infusion may lead to increased duration of action and SE
- Remifentanil has little redistribution and a small Vd (0.2-0.3L/kg) in the peripheral compartments, and a very short elimination half life of 3 minutes. Hence, it has a very short, context-insensitive half time
It wears off reliably and quickly following cessation of infusion.

Courtesy: icuprimaryprep.com
Examiner Comments
2024A 09: 31% of candidates passed this question.
This question is essentially a compare and contrast question. It was worded specifically to encourage candidates to describe the pharmacokinetics of the drugs in question (in particular the relative lipid solubilities, volume of distribution, pka/ionisation, plasma protein binding, and metabolism) and then use these features to discuss the clinically relevant implications. In part (c) a brief definition of what is meant by the context sensitive half time followed by how these pharmacokinetic differences influence the CSHT of each was required.
2018A 05: 66% of candidates passed this question.
Well-constructed answers were presented in a table to compare pharmacokinetics and pharmacodynamics with a separate paragraph to discuss the concept of context sensitive halftime. Important pharmacokinetic points included: the differences in lipid solubility, ionised fractions and onset, and differences in metabolism. Marks were awarded for a definition of context-sensitive half-time. A discussion of these two drugs’ context-sensitive half-times should have included the differences in re-distribution into other compartments and rates of elimination.
2017B 13
Compare and contrast the pharmacology of intravenous fentanyl and morphine
Examiner Comments
2017B 13: 68% of candidates passed this question.
Good candidates produced a well-structured answer and highlighted the differences between the two drugs. It was important to include the dose, potency, time course of effect of both agents, and differences in pharmacokinetic and pharmacodynamic effects. Candidates should have specific knowledge of these important drugs. Many candidates failed to focus the question on intravenous fentanyl and intravenous morphine as asked. No marks were given for information about other routes of administration.
2016B 12
Compare and contrast the pharmacokinetics and adverse effects of morphine and fentanyl.
Examiner Comments
2016B 12: 20% of candidates passed this question.
This question is best answered in a tabular form, comparing absorption, distribution, metabolism, excretion and the adverse effects.
Common omissions were lack of details on distribution, and not relating lipid solubility to effect. A description of the relative adverse effects was expected, e.g., more histamine release, less bradycardia, rather than listing similar adverse effects, e.g. respiratory depression.
Comparisons of pharmaceutics and pharmacodynamics did not attract any additional marks.
2020A 17
Discuss the advantages and disadvantages of the use of an intravenous infusion of fentanyl in comparison to morphine.
CICMWrecks Answer
| Fentanyl Infusion | Morphine Infusion | |
|---|---|---|
| Cost | slightly less | |
| Potency | 50 to 100 times more potent highly lipophilic, which allows rapid penetration of the blood-brain barrier and rapid onset of action (four to six minutes), although maximal analgesic and respiratory depressant effects of fentanyl may not be evident for several minutes | |
| Effective analgesia for surgical trauma causing severe pain during the intraoperative or immediate postoperative period, due to a prolonged duration of action. | ||
| PD | a more rapid onset of action than morphine, which reflects its greater lipid solubility and consequent increased ability to cross the blood–brain barrier | Slower onset of analgesia (within 20 minutes) and slower time to peak analgesic effect compared with fentanyl due to lower lipid solubility and a longer lag time for penetration of the blood-brain barrier. |
| redistribution to inactive tissue sites such as fat and skeletal muscle, with an associated decrease in plasma concentration Furthermore, fentanyl has been reported to have 75% first-pass uptake into the lungs, thus the amount of fentanyl that reaches the systemic circulation is limited | morphine’s first-pass pulmonary uptake is extremely small | |
| Despite fentanyl being a more attractive option than morphine for analgo-sedation, because of its potentially shorter duration of action, other pharmacokinetic properties may limit its usefulness. After initial redistribution, fentanyl’s plasma concentrations are maintained by slow reuptake from the tissues across a concentration gradient | ||
| Effects | no significant difference in amount of pain relief | |
| Minimal effect on myocardial or hemodynamic function. | Unsuitable for patients with hemodynamic instability due to possible exacerbation of hypotension by histamine release, as well as persistence of its effects due to a long context-sensitive half time and the mu-receptor-stimulating properties of its morphine-6-glucuronide metabolite. | |
| Metabolism | Metabolised by N-demethylation in the liver, catalysed by the cytochrome p450 system; major metabolite is norfentanyl and other metabolites are hydroxyproprionyl-fentanyl and hydroxyproprionyl-norfentanyl (all these metabolites have minimal pharmacologic activity) | Metabolised by conjugation with glucuronic acid in hepatic and extrahepatic sites, especially the kidneys; major metabolites are morphine-3-glucuronide (75–85%), morphine-6-glucuronide (5–10%); other metabolites are normorphine and a small amount to codeine |
| Active metabolites | Minimal pharmacological effect | Morphine-6-glucuronide – pharmacologically active undergoes renal clearance and accumulates in patients with renal failure, with clinical effects including prolonged sedation and respiratory depression elimination half-life of about 1.4 hours, but this is increased in patients with renal failure |
| Fentanyl has a high hepatic extraction ratio with clearance approaching liver blood flow | ||
| Excretion | Metabolites are renally excreted and < 10% of fentanyl is excreted unchanged in the urine | Metabolites are renally excreted with 7–10% undergoing biliary excretion |
| Elimination half life | 3.1–6.6 hours | 1.7–2.3 hours |
| fentanyl’s effects may be as prolonged as morphine, even when allowing for potential accumulation of metabolically active metabolites of morphine in renal failure | Cautious use in patients with a history of seizures if renal insufficiency is present, due to neuroexcitation caused by the morphine-6-glucuronide metabolite, with possible occurrence of myoclonus or exacerbation of seizure activity. | |
| Context-sensitive half-time | Consequently, following infusion, the clinical effects of fentanyl become increasingly prolonged owing to its long “context-sensitive half-time” | |
CSHT 4hr inf | 200 minutes | Not applicable |
| CSHT 8hr inf | 300 min | Not applicable |
| longer inf | unpredictably long | Not applicable |
| Adverse effects | Higher incidence of most opioid-related adverse side effects (eg, pruritus, urinary retention, constipation, and nausea), compared with other opioids. | |
| Absence of histamine-releasing properties; thus, fentanyl is appropriate for patients with bronchospasm. | headedness, sedation, drowsiness and euphoria). In addition, morphine contains a tertiary amine group, which causes non-immune-mediated release of histamine and can lead to skin rashes, itchiness and hypotension | |
| Drug-drug interactions | Similar interactions – may enhance the serotonergic effect of Serotonergic Agents (High Risk). This could result in serotonin syndrome | |
Examiner Comments
2020A 17: 27% of candidates passed this question.
These are both level 1 drugs commonly used as an infusion in daily practice. This question specifically asked the candidates to frame their answers around an intravenous infusion of fentanyl in comparison to morphine. A tabular listing of general properties of the two drugs highlighting the differences between the drugs would not score well. The question asks for a considered response that should focus on context sensitive half-life, compartments and metabolism, instead many focused on the speed of onset and potency, which are minor considerations when drugs are given for long periods by infusion. Candidates often demonstrated a superficial knowledge of key pharmacokinetic concepts with limited application of these principles in the context of an intravenous infusion. Better answers also related the above to various relevant pharmacodynamic influences such as age, liver and renal impairment.
2011A 16
Compare and contrast the pharmacology of morphine, fentanyl and remifentanil.
Examiner Comments
2011A 16: 2 (17%) of candidates passed this question.
The question asked for a comparison of the pharmacology (pharmacokinetics and pharmacodynamics) of three commonly used opiates. Better answers made use of a well constructed table with headings including chemistry, protein binding, lipid solubility, half-lives, context sensitive half-time, volume of distribution, metabolism, active metabolites, oral bioavailability, and clearance. A distinction should have been clearly drawn between onset, peak, and duration of effect. CNS stimulant effects as well as depressant effects, were expected to be listed.
Syllabus: G2d, 2d
Recommended sources: Anaesthesia, Miller Chp 11 and Pharmacological Basis of Therapeutics, Goodman and Gillman, Chp 21
2013B 14
Compare and contrast the mechanism of action, pharmacokinetics and central nervous system effects of morphine and tramadol.
Examiner Comments
2013B 14: 15 candidates passed (55.6%).
In general candidates either lacked a depth of knowledge, or a deep enough understanding of the drugs so as to apply their knowledge specifically to the central nervous system (CNS). Mechanism of action and pharmacokinetics for morphine was better understood, in comparison to tramadol. Mention of the non-CNS effects of morphine and tramadol, was not expected, did not score marks, and would have wasted valuable exam time.
2021A 12 – 2017A 12
Describe the pharmacology of oxycodone.
Examiner Comments
2021A 12: 54% of candidates passed this question.
There were many exceptional answers which provided extensive detail on the drug. The best of these gave context for the drug characteristics, such as by referring to oxycodone relative to other opioid drugs that might be chosen, or to considerations for safe and effective administration. Some answers, however, provided generic information on opioid drugs, which could not gain all the available marks.
2017A 12: 53% of candidates passed this question.
Few candidates covered the pharmacokinetic aspect of the question sufficiently. No marks were awarded for generic comments such as hepatic metabolism and renal excretion.
2009A 12
Outline the pharmacology of an opioid injected into the spinal intrathecal space.
CICMWrecks Answer
Pharmacokinetic Effects
- D: Dependent on degree of lipid solubility. Fentanyl has > systemic absorption than Morphine. More likely to cause systemic opoid effects
- M: Minimal metabolic effects occur in the intrathecal space
- E: Clearance from the intrathecal space is via the arachnoid villi.
Pharmacodynamic Effects and Side-effects
Secondary to:
- Local Effects
- Analgesia
- MOP in spinal cord → ↓pain transmission primary afferent neurons
- Orthostatic hypotension
- 2° direct SNS blockade at sympathetic chain → vasodilation → venous pooling
- ↓ temperature
- Inhibition of shivering
- Analgesia
- Cephalic migration:
- Depends primarily on the lipid solubility of the drug
- Sedation
- Direct effect on μ-receptors in reticular formation
- Confusion
- Anticholinergic (pre-synaptic blockade of ACh release)
- Pruritis
- Interaction with μ-receptors in trigeminal nucleus
- CNS Excitation
- In overdose → myoclonic jerks
- Due to bulk flow to brainstem/basal ganglia
- ↓MV
- Direct opioid effect depressing ventilatory centre of brainstem
- ↑PaCO2 due to right-shift in ventilation v PaCO2 curve
- N&V
- Systemic Absorption
- ↓gastric emptying, ↓gut motility
- ↓MAP
- Mast cell degranulation → histamine release → vasodilation (rare with fentanyl, ↑with morphine)
Gladwin 2016
Examiner Comments
2009A 12: Pass rate: 30%
Though it would be unusual for patients to receive spinal opioids whilst they are in intensive care, the complications of spinal opioids are not an uncommon reason for admission to intensive care thus it is important candidates understand their pharmacology. Answers generally lacked structure. Outlining pharmacology should include pharmacokinetics, pharmacodynamics and side effects (both common and dangerous). An explanation of the effect of lipid solubility was expected. Following a structure will ensure a more complete answer.
Syllabus G2d2e
Reference: Neural Blockade. 3rd edition Cousins and Bridenbaugh
Non-Opiates
2014A 05 – 2008A 01
Describe the pharmacological effects of paracetamol. (40% of marks)
Outline its toxic effects and their management. (60% of marks)
CICMWrecks Answer
Pharmacological effects of Paracetamol
- Arachidonic acid ——(COX)—–>
- Prostaglandins → inflammation, sensitise spinal nerves to pain
- Prostacycline → vasodilation
- Thromboxane → platelet aggregation and activation
- Paracetamol: Weak COX-1 and COX-2 inhibitor in peripheral tissues
- Relatively selective for COX-2 → less inhibition of thromboxane production
- Possible cause for antipyretic effect:
- Inhibition of COX-3 (variant of COX-1), leading to central effects
Toxic Effects and Management
Toxicity
- Occurs in doses >4g/day
- Overdose dose lower in chronic alcoholics, elderly and children
- Paracetamol
- → glucuronidation
- → sulphation
- → N-hydroxylation to NAPQI
- In overdose, glucuronidation and sulphation pathways saturated → increased production of toxic NAPQI
- NAPQI usually conjugated with hepatic glutathione
- In overdose, glutathione exhausted → accumulation of NAPQI
- NAPQI bonds with hepatocyte sulfhydryl groups → cell death and necrosis
Therapy
- Decision to treat based on paracetamol overdose nomogram
- Serum paracetamol level, time since dose
- If within 4 hours → activated charcoal
- Hepatic failure, GI upset, renal failure (less common)
- N-acetyl-cysteine should be given
- Replenishes glutathione stores
- Directly conjugates NAPQI
- Monitoring and supportive care:
- Bilirubin
- Transaminases
- Creatinine (dialysis)
- Liver synthetic function (albumin, INR) (vitamin K, maybe FFP, but discuss with liver unit first)
- Blood sugar (glucose infusion)
- Conscious level (hepatic encephalopathy) (lactulose, intubation)
Mooney 2016
Examiner Comments
2014A 05: 63% of candidates passed this question.
This question was generally well answered with narrow variance; very few candidates discussed factors predisposing to hepato-toxicity or renal toxicity. Discussion of pharmacokinetics only gained marks when relevant to toxicity.
2008A 01: 2 candidates (66%) passed this question.
Main points expected for a pass included:
- Actions and mechanism of actions of paracetamol. These include inhibition of cylooxygenase (COX) activity and prevention of prostaglandin (PG) production. More marks were given for mention of a central COX 1 variant as the possible enzyme responsible for paracetamol’s central versus peripheral effects. Knowledge of the central sites of action was expected.
- Outline of toxicity. Candidates were expected to demonstrate knowledge of toxic doses, conditions enhancing toxicity (alcohol intake, chronic use etc), and the mechanism of hepatic toxicity and other organ toxicity (especially renal). A detailed list of clinical features of toxicity was not required.
- Management of toxicity. Candidates performed well in this section with good knowledge of timing, toxic doses, use of paracetamol levels and the nomogram to determine whether N-acetylcysteine should be administered. Mechanism of action of NAC was expected. Mention of monitoring (liver failure) and supportive therapy gained marks, but detailed explanations were not required
Syllabus G2e2c
Stoelting 4th edition page 285
Rand Dale 5th page 244
2011B 12
List the functions of the liver (60% marks). Discuss the metabolism of paracetamol in toxicity and the pharmacologic management of this overdose (40% marks).
CICMWrecks Answer: Liver Functions
Liver:
- Largest abdominal solid organ
- 25 % CO at rest. approx 1,500 mls/min
- 15% of total blood volume at rest
Functions of Liver
| Filtration | |
| Immune defence | (via Kuppfer cells) against agents entering the portal circulation |
| 80% of circulating cholesterol | → bile salt |
| Biliary excretion of drugs/hormones | penicillins, amp, erythro |
| thyroxine, cortisol, estrogen | |
| calcium | |
| Immune | |
| Filtration of portal circulation | |
| Kuppfer cells | bacteria/ virus/ endotoxins/ immune complexes/ thrombin/ tumour |
| Phagocytosed, fused with lysozomes and degraded by lysosomal enzymes | |
| Antigen presentation | |
| Endotoxin neutralisation = pinocytosed | |
| Complement/CRP production | |
| Storage of metabolic substrate/fluids | |
| glycogen ~ 400g | |
| fat | |
| Fe++, B12, folate, Cu | |
| Vitamin A | |
| Blood Reservoir | |
| Metabolic | |
| CHO and intermediary metabolism | Hepatic Glucostat |
| gluconeogenesis, glycogen storage and utilisation, galactose/fructose to glucose | |
| Conversion to fat, AA’s and ketones | |
| Protein metabolism | amino acids utilisation |
| protein synthesis | |
| production of ketones | |
| deamination of fatty acids | |
| urea formation for ammonia removal | |
| plasma protein formation | |
| Fat homeostasis | metabolism (beta oxidation (rapid in hepatic cells)), synthesis and transport as lipoprotein |
| cholesterol homeostasis | |
| Endocrine | hormone synthesis & metabolism |
| Synthesis of 25 OH cholecalciferol, Metabolism of steroid hormones, Synthesis of somatomedins, Erythropoietin | |
| biotransformation | ammonia & urea cycle |
| drugs & toxins | |
| Acid-Base | Lactate metabolism |
| Synthetic functions | |
| Bile production | bile salts (incr fat absorption) |
| bilirubin (incr haem excretion) | |
| Protein synthesis | albumin 120-300mg/kg/d |
| alpha1/2 & beta globulins (transport) | |
| coagulation & fibrinolytic factors (fibrinogen, prothrombin, II, V, VII,VII, IX, X, XI, XII, XIII, antithrombin) | |
| Lymph synthesis | Up to 50% |
| Erythropoietin (10%) | |
Gladwin / JC / Bianca 2019
CICMWrecks Answer: Paracetamol Toxicity
Paracetamol Overdose
Hepatotoxicity
Toxic daily dose > 4-6 g (while lethal dose occurs at > 10-15 g (or > 300 mg/kg LBM)) → but the toxic and lethal doses are lower in “at-risk” groups:
- EtOH abuse (due to CYP450 induction (↑ toxic metabolite produced) and ↓ glutathione stores)
- Malnutrition (due to ↓ glutathione stores)
- Elderly (due to ↓ glutathione stores)
- Preexisting liver dysfunction
Mechanism:
- At therapeutic doses – “N-acetyl-p-amino-benzoquinoneimine” (highly toxic metabolite) is produced in small amounts, but is rapidly conjugated with hepatic glutathione (anti-oxidant) into a harmless metabolite
- BUT with toxic doses – Hepatic conjugation pathway is saturated and ↑↑↑ N-acetyl-p-amino-benzoquinoneimine is produced → this depletes hepatic glutathione stores, causing remaining N-acetyl-p-aminobenzoquinoneimine to then forms covalent bonds with sulphydryl groups on hepatocytes → results in centrilobular hepatic necrosis
Clinical features:
- Generally conscious and c/o N/V, epigastric pain, erythema, sweating → later developing:
- Acute haemolytic anaemia
- Develop hepatic failure (Ie. jaundice and cholestasis) after 48 hrs
- LFT/INR derangements at 3-5 days
- Fulminant hepatic failure at 3-7 days
- With severe OD, can present with hypotension/shock
Diagnosis :
Serum paracetamol levels → correlate with “nomogram” to predict likelihood of liver damage → used as a guide to dictate therapy
Treatment:
- Activated charcoal/gastric lavage→ limit paracetamol absorption
- Replace hepatic glutathione store within 12 hrs of OD → permits glucuronidation of toxic metabolite
- Oral methionine → ↑ glutathione synthesis
- IV N-acetylcysteine → hydrolysed to cysteine (which is a glutathione precursor)
- Nb. IV NAC is preferred b/c of N/V a/w toxicity (Ie. ↓ oral methionine absorption)
- IV glucose → due to risk of ↓ BGL with liver dysfunction
- Serial monitoring of LFTs and coagulation studies
- Referral to a specialist centre
Gladwin / JC / Bianca 2019
Examiner Comments
2011B 12: 5 (20%) of candidates passed this question.
A good response to this question required an ordered and well structured response. There are numerous important functions that the liver undertakes (eg bile formation, immunological, protein, lipid, glucose metabolism, storage, endocrine, etc), yet most candidates could not generate a sufficient list. In relation to paracetomol toxicity, a good response required candidates to mention that normal conjugating reactions in the liver are saturated and metabolism diverts to mixed function oxidases, which generate toxic metabolites. These in turn are inactivated by conjugation with glutathione. However when glutathione is depleted, toxic metabolites react with cellular nuclear material, thus causing liver necrosis. Toxic compound depletes sulphydril groups and also causes direct damage via lipid peroxidation. Regeneration of sulphydril groups and glutathione depends on availability of cysteine, thus the need to administer acetylcysteine.
Syllabus: I2a, G2e2c
Recommended sources: Gannong Review of Medical Physiology pg 485; Power and Kam
Principles of Physiology for the Anaesthetist pg185; Stoelting Pharmacology and Physiology in Anesthetic Practice 4th edition pg 285; Rang & Dale Pharmacology 5th Ed pg 244.
2017B 01
Compare and contrast the pharmacology of ibuprofen and paracetamol
Examiner Comments
2017B 01: 65% of candidates passed this question.
This was a standard compare and contrast question of common analgesic pharmacology and it was generally well answered. The use of a table ensured all areas were covered including class, indications, pharmaceutics, mode of action, pharmacodynamics, pharmacokinetics and adverse effects. The uncertain nature (and possibilities) of the mechanism of action of paracetamol was alluded to in better responses.
Details of the comparative pharmacokinetics were often lacking. Answers should have included a comment on first-pass effect, the significance of the difference in protein binding and the details of metabolism, particularly paracetamol. Metabolism limited to “hepatic metabolism and renal excretion” gained no marks as better responses were more detailed and clearer about the differences between the two drugs. Knowledge of metabolism at therapeutic doses and the effect of overdose were expected. Better answers included potential interactions with other drugs (e.g. warfarin) and contraindications to the use of these drugs.
2007B 15
Compare and contrast ibuprofen and tramadol as analgesic agents in intensive care.
Examiner Comments
2007B 15: 3 candidates (43%) passed this question.
Ibuprofen – inhibition of cyclooxygenase (COX) and synthesis of prostaglandins, which are important mediators for peripheral sensitization and hyperalgesia. Act peripherally and spinal COX – non selective. Oral and PR only. Associated with a number of side effects, including decreased haemostasis, renal dysfunction, gastrointestinal hemorrhage, and effects on bone healing and osteogenesis.
Tramadol – is a synthetic opioid that exhibits weak μ-agonist activity and inhibits reuptake of serotonin and noradrenaline. Analgesic effects primarily through central mechanisms, it may exhibit peripheral local anaesthetic properties.
Tramadol is comparable in analgesic efficacy to ibuprofen. Common side effects (overall incidence of 1.6% to 6.1%) include dizziness, drowsiness, sweating, nausea, vomiting, dry mouth and headache. Tramadol should be used with caution in patients with seizures or increased intracranial pressure and in those taking monoamine oxidase inhibitors. IV and oral preparations. No bleeding, GIT and renal complications. More expensive.
Both have advantage of lack of respiratory depression, major organ toxicity, and depression of gastrointestinal motility and a low potential for abuse.
NMDA Antagonists
2022A 11
Outline the structure and function of the N-methyl-D-aspartate (NMDA) receptor (25% marks). Discuss the pharmacology of ketamine (75% marks).
2019A 15
Describe the physiology of the NMDA (N-Methyl D-aspartate) receptor (40% of marks).
Outline the pharmacology of ketamine (60% of marks).
CICMWrecks Answer: NMDA Receptor
N-Methyl D-Aspartate (NMDA) Receptor
- ligand-gated voltage dependent ion channel
- Natural ligand = glutamate
- Associated with the family of glutamate receptors (AMPA, kainite and neurokinin)
Structure:
- transmembrane receptor
- 5 subunits forming central cation ionophore
- at baseline Mg2+ plugs and inhibits central cation pore
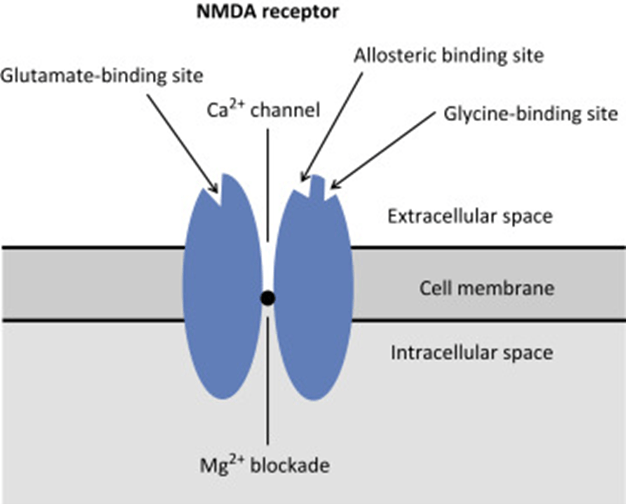
- Location: abundant throughout brain (esp. hippocampus) and spinal cord (esp. dorsal horn) on post-synaptic membranes
- Activation: NMDA receptor contains central Mg2+ plug, which block channel at rest
- Glycine binding + voltage stimulus are required (via activation of adjacent AMPA and neurokinin receptors) → remove Mg plug → primes NMDA receptor for activation by glutamate
- When channel activated→ opens cation channel → ↑cation conductance (Ca2+ and Na+ in; K+ out) → excitatory postsynaptic potential
Physiological Roles of NMDA Receptors in the CNS
- Wind up phenomenon
- Wind up = repeated stimuli of same strength cause increase in pain response
- Proposed mechanism: C fibres synapse in lamina II of dorsal horn → repeated stimulation → glutamate release → NMDA activation → ↑response of dorsal horn neurons to excitatory neurotransmitter input àhyperalgesia and allodynia
- Long term potentiation
- LTP = strengthening of synaptic transmission that occurs following ↑activity across that synapse
- i.e. recurrent painful stimuli → neuroplasticity → chronic pain
- NMDA receptor stimulation leads to various intracellular changes:
- production of NO
- activate second messengers (IP3, DAG, cGMP, PKC)
- PKC → increases NMDA activity → vicious cycle
- second messengers induce oncogenes (e.g. c-fos)
- Memory
- NMDA receptors are highly expressed in hippocampus and throughout cortex Important roles in neuroplasticity and memory formation
- Apoptosis
- Post cerebrovascular accident → significant NMDA activation → ↑↑Ca influx → excitotoxicity → triggers neuronal apoptosis
CICMWrecks Answer: Ketamine Pharmacology
Pharmacology of Ketamine
Examiner Comments
2022A 11: 74% of candidates passed this question.
The first part of this question required a description of both the receptor structure and its function. This includes, but is not limited to, its location, the natural ligand, how the channel may be regulated and the results of receptor stimulation. The second part of this question related to ketamine. Marks lost here often related to vague statements and incorrect facts. The examiners also commented that some candidates got confused between the R and S enantiomers. Few candidates commented on the nature of the metabolites and generally the PD section was vaguely answered.
2019A 15: 49% of candidates passed this question.
The NMDA receptor is a ligand gated voltage dependent ion channel located on post synaptic membranes throughout the CNS, with glutamate, an excitatory neurotransmitter, its natural ligand. A brief description of its structure, roles of glycine and magnesium, ions conducted, result of activation, role in memory and learning and agonists/antagonists was expected. Detail on structure and functions of the receptor were a common omission.
Ketamine, a phencyclidine derivative, is a non-competitive antagonist at the NMDA receptor. It is presented as a racemic mixture or as the single S(+) enantiomer (2-3 X potency).
Administration routes and doses scored marks. Pharmacodynamics were generally well covered including CVS (direct and indirect effects), CNS (anaesthesia, analgesia, amnesia, delirium, effects on CBF and ICP) respiratory (bronchodilator with preservation of airway reflexes) GIT effects (salivation, N and V). Knowledge of specific pharmacokinetic parameters was less well covered, including low oral bioavailability and protein binding and active metabolite (norketamine).
Antidotes
2013A 15
Describe the pharmacology of naloxone.
Examiner Comments
2013A 15:
Naloxone is a commonly used intravenous opioid antagonist, which acts as a competitive antagonist with high affinity for the mu, kappa, delta and sigma opioid receptors. It is used to ameliorate or reverse opioid effects at these sites. It has a shorter effect site and plasma half-life than most opiates so levels will fall before the opioid agonist it is being used to treat, thus a repeat dose maybe required to maintain opioid reversal. Overall candidates lacked sufficient depth of information to achieve high marks for this question.
ii. Outline the classification, structure and distribution of opioid receptors and NMDA receptors.
2023B 09
Outline the classification, structure and distribution of the opioid receptors (50% marks). Describe the intracellular events following opioid receptor activation (50% marks).
CICMWrecks Answer
Opioid Receptors
- Serpentine, inhibitory G protein-coupled receptors (GiPCR)
- ligands are endogenous opioids
- dynorphins, enkephalins, endorphins, endomorphins and nociceptin
- 4 types – MOR (), DOR (), KOR (), NOR
| Receptor | Subtypes | Location | Clinical Effects | ||
|---|---|---|---|---|---|
| Brain | Spinal Cord | Other | |||
| MOR (μ) | μ1, μ2, μ3 | cortex (laminae III and IV) thalamus striosomes periaqueductal gray rostral ventromedial medulla | substantia gelatinosa | peripheral sensory neurons intestinal tract | μ1: analgesia physical dependence μ2: respiratory depression miosis euphoria reduced GI motility physical dependence μ3: possible vasodilation |
| DOR (δ) | δ1, δ2 | pontine nuclei amygdala olfactory bulbs deep cortex | – | peripheral sensory neurons | Analgesia Antidepressant Convulsant Physical dependence May modulate MOR mediated respiratory depression |
| KOR (κ) | κ1, κ2, κ3 | hypothalamus periaqueductal gray claustrum | substantia gelatinosa | peripheral sensory neurons | Central analgesia anticonvulsant, depression, dissociative, hallucinogenic diuresis, miosis, dysphoria sedation, stress Visceral nociception antagonist |
| NOR (Nociceptin) | ORL1 | cortex amygdala hippocampus septal nuclei habenula hypothalamus | spinal cord | – | anxiety, depression appetite development of tolerance to μ-opioid agonists |
Opioid Receptor Structure and Activation
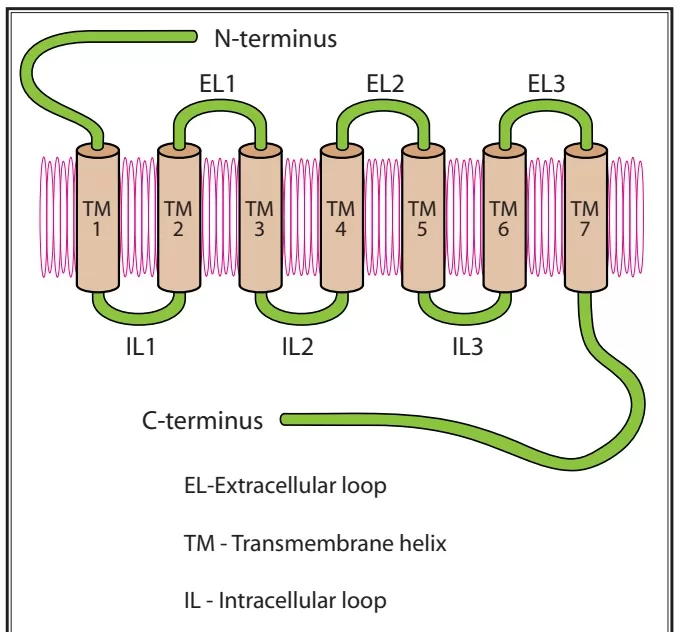
- Each receptor consists of an extracellular N-terminus, 7 transmembrane helical twists, 3 extracellular and intracellular loops, and an intracellular C-terminus
- Once the receptor is activated, it releases a portion of the G protein, which diffuses within the membrane until it reaches its target (either an enzyme or an ion channel).
- These targets alter protein phosphorylation via inhibition of cyclic AMP (cAMP) which acts as a second messenger within the cell resulting in
- the activation of protein kinases (short term effects) and
- gene transcription proteins and/or gene transcription (long term effects)
- Neurotransmitter release from neurons is normally preceded by depolarisation of the nerve terminal and Ca++ entry through voltage-sensitive Ca++ channels.
- Opioid receptors located on the presynaptic terminals of the nociceptive C-fibers and A delta fibers, when activated by an opioid agonist, will indirectly inhibit these voltage-dependent calcium channels →
- decreasing cAMP levels and blocking the release of pain neurotransmitters such as glutamate, substance P, and calcitonin gene-related peptide from the nociceptive fibers →
- resulting in analgesia
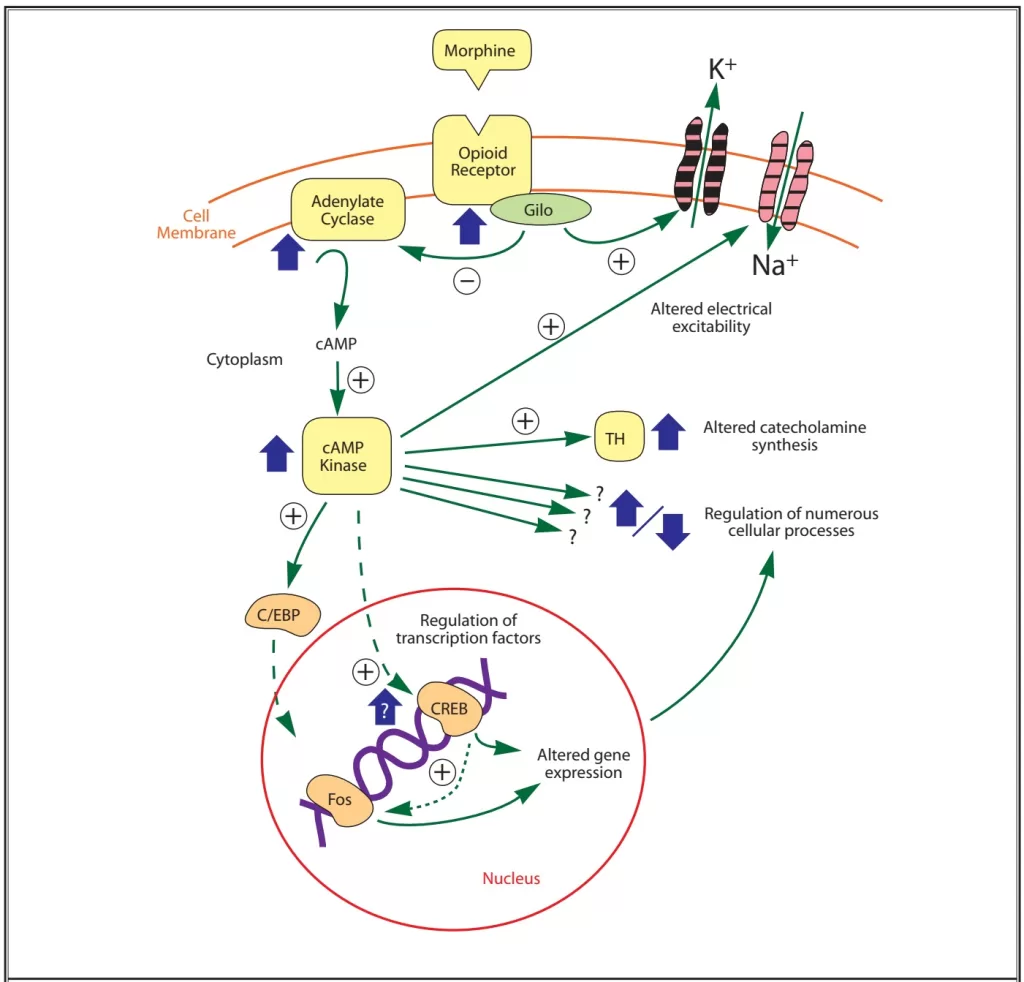
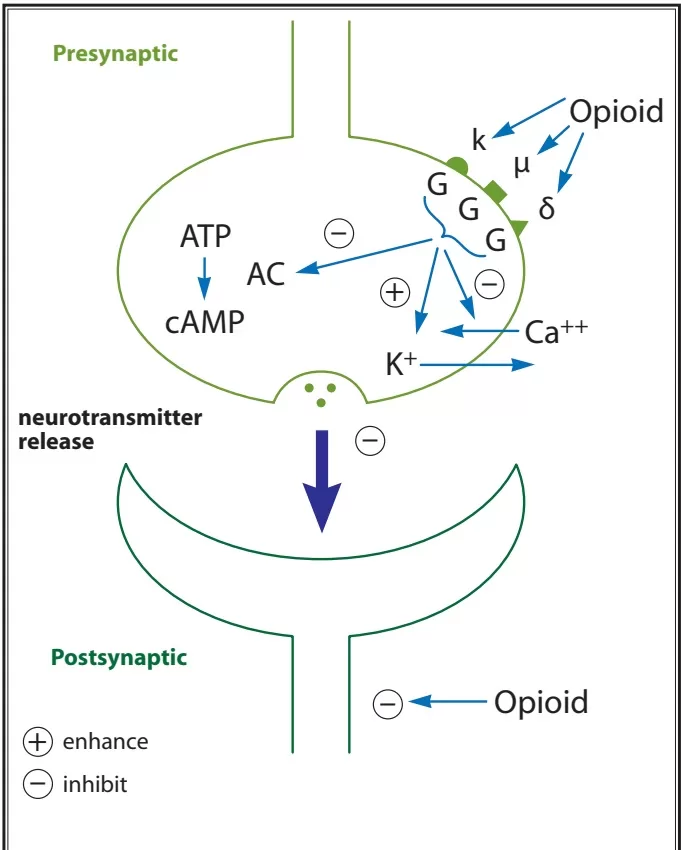
- Opiod receptor activation → Increases adenylyl cyclase, reducing cAMP
- Presynaptically inhibits voltage-gated Ca2+ channels
- Decreases Ca2+ influx
- Reduces neurotransmitter release
- Post-synaptically activates K+ channels
- Causes K+ efflux
- Leads to membrane hyperpolarisation
- Presynaptically inhibits voltage-gated Ca2+ channels
- Overall this leads to a reduction in neuronal cell excitability, resulting in reduced transmission of nociceptive impulses.
Sources: Notes,
https://www.painphysicianjournal.com/current/pdf?article=OTg3&journal=42
Examiner Comments
2023B 09: 20% of candidates passed this question.
This question was asked in a specific way to provide candidates with a template for their answer. The classification most commonly used for opioid receptors (μ(MOP), δ(DOP), k(KOP) & NOP) and a description of the important characteristics or differences between them was expected. A description of their central and peripheral distribution was required including specific central nervous system sites such as pre and post synaptic locations in the brain (ie. the periaqueductal gray, locus ceruleus and amygdala) and spinal cord (ie. primary afferent neurons in the dorsal horn).
Opioid receptors as a class are transmembrane spanning G protein receptors that have significant downstream effects including presynaptic inhibition of neurotransmitters of primary afferent neurons such as noradrenaline and substance P, and postsynaptic inhibition of membrane depolarization of dorsal horn nociceptive neurons. Specifity of detail in descriptions of these actions was expected.
2022A 11
Outline the structure and function of the N-methyl-D-aspartate (NMDA) receptor (25% marks). Discuss the pharmacology of ketamine (75% marks).
2019A 15
Describe the physiology of the NMDA (N-Methyl D-aspartate) receptor (40% of marks).
Outline the pharmacology of ketamine (60% of marks).
CICMWrecks Answer: NMDA Receptor
N-Methyl D-Aspartate (NMDA) Receptor
- ligand-gated voltage dependent ion channel
- Natural ligand = glutamate
- Associated with the family of glutamate receptors (AMPA, kainite and neurokinin)
Structure:
- transmembrane receptor
- 5 subunits forming central cation ionophore
- at baseline Mg2+ plugs and inhibits central cation pore
- Location: abundant throughout brain (esp. hippocampus) and spinal cord (esp. dorsal horn) on post-synaptic membranes
- Activation: NMDA receptor contains central Mg2+ plug, which block channel at rest
- Glycine binding + voltage stimulus are required (via activation of adjacent AMPA and neurokinin receptors) → remove Mg plug → primes NMDA receptor for activation by glutamate
- When channel activated→ opens cation channel → ↑cation conductance (Ca2+ and Na+ in; K+ out) → excitatory postsynaptic potential
Physiological Roles of NMDA Receptors in the CNS
- Wind up phenomenon
- Wind up = repeated stimuli of same strength cause increase in pain response
- Proposed mechanism: C fibres synapse in lamina II of dorsal horn → repeated stimulation → glutamate release → NMDA activation → ↑response of dorsal horn neurons to excitatory neurotransmitter input àhyperalgesia and allodynia
- Long term potentiation
- LTP = strengthening of synaptic transmission that occurs following ↑activity across that synapse
- i.e. recurrent painful stimuli → neuroplasticity → chronic pain
- NMDA receptor stimulation leads to various intracellular changes:
- production of NO
- activate second messengers (IP3, DAG, cGMP, PKC)
- PKC → increases NMDA activity → vicious cycle
- second messengers induce oncogenes (e.g. c-fos)
- Memory
- NMDA receptors are highly expressed in hippocampus and throughout cortex Important roles in neuroplasticity and memory formation
- Apoptosis
- Post cerebrovascular accident → significant NMDA activation → ↑↑Ca influx → excitotoxicity → triggers neuronal apoptosis
CICMWrecks Answer: Ketamine Pharmacology
Pharmacology of Ketamine
Examiner Comments
2022A 11: 74% of candidates passed this question.
The first part of this question required a description of both the receptor structure and its function. This includes, but is not limited to, its location, the natural ligand, how the channel may be regulated and the results of receptor stimulation. The second part of this question related to ketamine. Marks lost here often related to vague statements and incorrect facts. The examiners also commented that some candidates got confused between the R and S enantiomers. Few candidates commented on the nature of the metabolites and generally the PD section was vaguely answered.
2019A 15: 49% of candidates passed this question.
The NMDA receptor is a ligand gated voltage dependent ion channel located on post synaptic membranes throughout the CNS, with glutamate, an excitatory neurotransmitter, its natural ligand. A brief description of its structure, roles of glycine and magnesium, ions conducted, result of activation, role in memory and learning and agonists/antagonists was expected. Detail on structure and functions of the receptor were a common omission.
Ketamine, a phencyclidine derivative, is a non-competitive antagonist at the NMDA receptor. It is presented as a racemic mixture or as the single S(+) enantiomer (2-3 X potency).
Administration routes and doses scored marks. Pharmacodynamics were generally well covered including CVS (direct and indirect effects), CNS (anaesthesia, analgesia, amnesia, delirium, effects on CBF and ICP) respiratory (bronchodilator with preservation of airway reflexes) GIT effects (salivation, N and V). Knowledge of specific pharmacokinetic parameters was less well covered, including low oral bioavailability and protein binding and active metabolite (norketamine).
Recent Comments