Syllabus (Fourth Edition, 2023)
Topics
i. Understand the pharmacology of sedating drugs.
ii. Understand the pharmacology of local anaesthetic drugs.
iii. Understand the pharmacology of anti-convulsant drugs.
iv. Understand the pharmacology of anti-depressant and anti-psychotic drugs.
Topics not covered in previous SAQs
.
Learning Objectives for the First Part Examination in Intensive Care Medicine
- This will ensure that trainees, tutors, and examiners can work from a common base.
- All examination questions are based around this Syllabus.
- These learning objectives are designed to outline the minimum level of understanding required for each topic.
- The accompanying texts are recommended on the basis that the material contained within them provides sufficient information for trainees to meet the learning objectives.
- Trainees are strongly encouraged to explore the existing and evolving body of knowledge of the Basic Sciences as they apply to Intensive Care Medicine by reading widely.
- For all sections of the syllabus an understanding of normal physiology and physiology at extremes of age, obesity, pregnancy (including foetal) and disease (particularly critical illness) is expected.
- Similarly, for pharmacology, trainees are expected to understand a drug’s pharmacology in these contexts.
- An understanding of potential toxicity and relevant antidotes is also expected.
Definitions
Throughout the document specific wording has been used under the required abilities to indicate the level of knowledge and understanding expected and a glossary of these terms is provided.
Definitions
| Calculate | Work out or estimate using mathematical principles. |
| Classify | Divide into categories; organise, arrange. |
| Compare and contrast | Examine similarities and differences. |
| Define | Give the precise meaning. |
| Describe | Give a detailed account of. |
| Explain | Make plain. |
| Interpret | Explain the meaning or significance. |
| Outline | Provide a summary of the important points. |
| Relate | Show a connection between. |
| Understand | Appreciate the details of; comprehend. |
SAQs
i. Understand the pharmacology of sedating drugs.
2016A 24
Describe the ideal sedative agent for an Intensive Care patient (50% of marks). How does midazolam compare to this (50% of marks)?
2008B 10
Outline the pharmacological properties of an ideal agent for sedating patients undergoing mechanical ventilation in intensive care (50% of marks). Describe how propofol compares to the ‘ideal’ agent (50% of marks).
CICMWrecks Answer
PHARMACEUTIC
| IDEAL SEDATIVE | MIDAZOLAM Y/N | PROPOFOL Y/N |
|---|---|---|
| long shelf life | Y | Y |
| stable when drawn up and on exposure to light | Y | Y |
| water soluble | Y | N |
| no refridgeration | Y | Y |
| cheap | Y | Y |
| mixes well with other agents in the central line lumen | Y | Y |
| Bacteriostatic | Y | N |
PHARMACODYNAMIC
| IDEAL SEDATIVE | MIDAZOLAM Y/N | PROPOFOL Y/N |
|---|---|---|
| Known MOA | Y | N |
| Affects only CNS | N | N |
| No excitatory or emergence phenomenon | Y | N |
| No pain on injection | rare | N |
| Safe on Intra-arterial injection | N | N |
| No trigger for MH or porphyria | Y | N |
| Safe in pregnancy | N | Y |
| Reliable dose – effect curve with little inter- individual variation in effect | N | Y |
| Anxiolysis | Y | Y |
| Analgesic properties | N | N |
| Amnesia | Y | Y |
| No effect on cardiovascular performance | N ↓MAP, ↓SVR, ↓HR | N ↓MAP, ↓SVR, ↓HR |
| Does not depress respiratory drive | N Significant, Dose-dependent | N Mild, dose-dependent |
| Rousable sedation | Y | Y |
| Minimal side effects | N | N |
| No incidences of allergy / anaphylaxis | Y rare | Y rare |
| No idiosyncratic reactions | N | N |
| No tachyphylaxis | N | Y |
PHARMACOKINETIC
| IDEAL SEDATIVE | MIDAZOLAM Y/N | PROPOFOL Y/N |
|---|---|---|
| Low volume of distribution | Y 0.8-1.5L/kg | N 2-10 L/kg |
| rapid onset | Y 2 mins | Y 30sec |
| low protein binding | N 96% PB | N 98% PB |
| Largely unionized (high pKa) | Y pKa=6.5 (85% ionized at 7.4) | Y pKa=11 (99% unionized at 7.4) |
| Lipophilic | Y Highly lipophilic with closed ring | Y |
| inactive metabolites | N | Y |
| non-toxic metabolites | Y | Y |
| rapid clearance (context-sensitive half-life) | Y/N (CSHT 40mins at 2hrs) | Y (CSHT 20mins at 2hrs) |
| clearance not affected by either renal or hepatic dysfunction | Y | N |
| Little inter-individual variation in pharmacokinetics | N | Y |
| Availability of an antagonist | Y Flumazenil | N |
Propofol infusion syndrome:
When used at >4mg/Kg/Hr for more than 24 hours
- Metabolic acidosis,
- Lipaemia,
- rhabdomyolysis
- Cardiac failure
- arrhythmias and death
Gladwin / JC 2020
Examiner Comments
2016A 24: 60% of candidates passed this question.
Candidates who had a structured approach (i.e. pharmaceutical, pharmacokinetic, pharmacodynamic) provided more content and scored higher. Candidates who also approached pharmacodynamic effects in an organ system based approach scored higher.
Relating a pharmacokinetic property of midazolam (e.g. volume of distribution or half-life) to a un/desirable attribute e.g. offset of action and accumulation displayed a greater understanding of the question. For many candidates, the description of an ideal drug contained more detail and candidates were not able to adequately state how midazolam compares.
2008B 10: 4 (80%) candidates passed this question
Candidates can benefit by having a system by which they approach topics that involve a broad and general topic such as that of the pharmacology of a particular drug or ideal agent.
A good answer included the following logical subheadings:
Desirable pharmacology – long shelf life, stable when drawn up and on exposure to light, cheap, mixes well with other agents in the central line lumen. Bacteriostatic.
Desirable pharmacokinetics – Low volume of distribution, rapid clearance (context-sensitive half-life), clearance not affected by either renal or hepatic dysfunction. Little inter-individual variation in pharmacokinetics. (Availability of an antagonist).
Desirable pharmacodynamics – Affects only CNS. Reliable dose – effect curve with little inter-individual variation in effect. Anxiolysis. (Analgesic properties). No effect on cardiovascular performance. Does not depress respiratory drive.
Minimal side effects – No incidences of allergy / anaphylaxis. No idiosyncratic reactions. No tachyphylaxis.
As indicated, 50% of the marks were allocated to mentioning how well propofol reflects these properties. Mention of ‘propofol infusion syndrome’ characterised by cardiac failure which can occur when propofol is used at >4mg/Kg/Hr for more than 24 hours also attracted marks.
Syllabus: G2a 1&2
Reference Text: Pharmacology and Physiology in Anesthetic Practice / R K Stoelting
2017B 11 – 2015B 14 – 2013B 21
Describe the pharmacology of propofol.
Examiner Comments
2017B 11: 76% of candidates passed this question.
A structured approach proved a good basis to answer this question. It was expected candidates would outline the uses such as anaesthesia, more prolonged sedation or possible additional roles in patients with seizures or head injuries. Discussion of the presentation and pharmaceutics, including a comment on antibacterial preservatives or lack thereof was expected. The mechanism of action should have been described. It was expected candidates could provide an indication of the usual dose (and how it differs in the more unwell / elderly patient population). A maximal rate and possible toxicity was expected.
A discussion on the pharmacodynamics by major organ systems was expected and credit was given for additional comments about hyperlipidaemia, urine colour changes or metabolic alterations. It was expected that candidates would mention propofol infusion syndrome at some point in their answer with some mention of clinical features or pathophysiology.
The important aspects of its pharmacokinetics should have been mentioned (high protein binding, large volume of distribution, termination of effect by redistribution, hepatic metabolism, context sensitive half life). A mention of adverse effects would complete the answer
2015B 14: 60% of candidates passed this question.
Those candidates who did poorly lacked any structure for answering a pharmacology question.
Pharmacokinetics was generally poorly handled and many answers revealed a lack of knowledge about this drug. Adverse effects and mechanism of action were generally well known. Doses of the drug were often incorrectly stated.
Important aspects such as dose or pharmacodynamics were often omitted and a structured approach helps avoid this
2013B 21: 19 candidates passed (70.4%).
A high level of knowledge was expected as it is a commonly used drug in intensive care. Overall most candidates performed very well. Areas of weakness were those relating to propofol pharmacokinetics and pharmacodynamics.
2022B 01
Describe the pharmacology of midazolam.
Examiner Comments
2022B 01: 88% of candidates passed this question.
Most candidates had a broad understanding of the indications, mechanism of action and the broad pharmacodynamic effects of midazolam. Those candidates who scored poorly demonstrated a lack of clarity and detail around the pharmacokinetics which are critical to its use and side effect profile. The examiners noted the details surrounding its bioavailability, protein binding was either almost universally absent or were generic statements that don’t score marks. Better candidates mentioned the changes in ring structure and lipophilicity associated with changes in pH and showed good understanding of clinically relevant facts regarding lipid solubility and active metabolites.
2019B 09
Compare and contrast the pharmacology of propofol and midazolam.
Examiner Comments
2019B 09: 77% of candidates passed this question.
Highlighting important similarities and differences between the drugs scored higher marks than listing the pharmacology of each drug separately. More pharmacokinetic information was required than simply stating both drugs “are metabolized in the liver and excreted by the kidney”.
2012A 05
Compare and contrast the pharmacology of dexmedetomidine and propofol.
Examiner Comments
2012A 05: 7 (70%) of candidates passed.
A basic and fundamental pharmacology question which required candidates to present their answer in a coherent fashion (a table worked best) as well as demonstrate sufficient knowledge. The majority of candidates did so, and so scored well. Candidates tended to struggle with the pharmacokinetic properties of these drugs.
2019A 07
Compare and contrast the pharmacokinetics and pharmacodynamics of midazolam and dexmedetomidine.
2008B 14
Compare and contrast the pharmacology of midazolam and dexmedetomidine when used for sedation.
Examiner Comments
2019A 07: 27% of candidates passed this question.
Most candidates used the effective tabular format presenting pharmacokinetics and pharmacodynamics of each drug side by side. Many answers demonstrated a lack of correct detail with respect to the pharmacokinetics and pharmacodynamics of these two level 1 drugs.
Many included pharmaceutics which attracted no marks as it was not asked.
2008B 14: 1 (20%) candidate passed this question
This question was also well suited to be answered in a preset format. For example a tabular format that had headings such as mechanism of action, preparations, dosing,
pharmacokinetics, metabolism and excretion, pharmacodynamics, drug interactions and side effects.
A good answer was expected to include the following points. Under mechanism of action, mention that both drugs produce sedation by hyperpolarizing CNS nerve membranes and act on different receptors (Midazolam binds the benzodiazepine receptor and dexmedetomidine being selective for the a2 receptor). Also mention of other effects for each drug, eg anxiolytic, anticonvulsunt, analgesia, etc. A similar approach would be required for other key areas such as metabolism and excretion,(including alterations with age, organ failure, disease, etc), drug interactions, pharmacodynamics, particularly in relation to important physiological effects (eg CNS and CVS effects).
A brief summary of the similarities and differences which influence the clinical use of these agents gained more marks and showed the candidate had applied knowledge of these drugs.
The common omissions were lack of explanation of mechanism of action and failure to mention pharmacodynamic effects, drug interactions and specific advantages for each agent.
Syllabus: G2b
2014B 21
Compare and contrast the pharmacology of haloperidol and diazepam.
Examiner Comments
2014B 21: 50% of candidates passed this question.
These are both commonly used agents and a tabulated format worked well. Subheadings covering the “general” pharmacology approach ensured core areas were addressed. Vague terms such as “good” or “moderate” did not allow a detailed comparison between the agents. Repetition of facts between sections such as uses, pharmacodynamics, effects and adverse effects did not gain further marks
2022A 11
Outline the structure and function of the N-methyl-D-aspartate (NMDA) receptor (25% marks). Discuss the pharmacology of ketamine (75% marks).
2019A 15
Describe the physiology of the NMDA (N-Methyl D-aspartate) receptor (40% of marks).
Outline the pharmacology of ketamine (60% of marks).
CICMWrecks Answer: NMDA Receptor
N-Methyl D-Aspartate (NMDA) Receptor
- ligand-gated voltage dependent ion channel
- Natural ligand = glutamate
- Associated with the family of glutamate receptors (AMPA, kainite and neurokinin)
Structure:
- transmembrane receptor
- 5 subunits forming central cation ionophore
- at baseline Mg2+ plugs and inhibits central cation pore
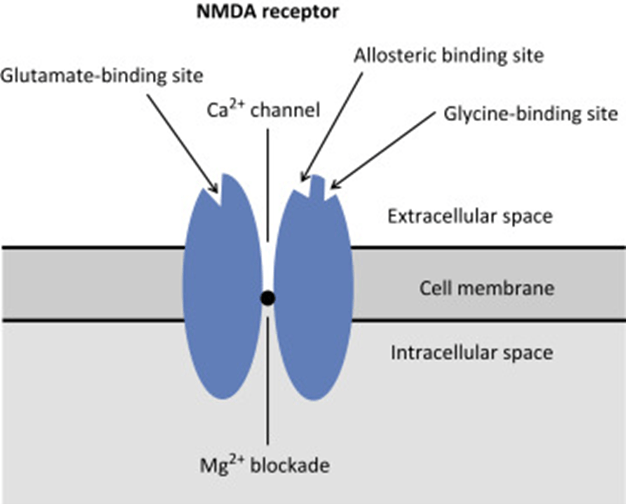
- Location: abundant throughout brain (esp. hippocampus) and spinal cord (esp. dorsal horn) on post-synaptic membranes
- Activation: NMDA receptor contains central Mg2+ plug, which block channel at rest
- Glycine binding + voltage stimulus are required (via activation of adjacent AMPA and neurokinin receptors) → remove Mg plug → primes NMDA receptor for activation by glutamate
- When channel activated→ opens cation channel → ↑cation conductance (Ca2+ and Na+ in; K+ out) → excitatory postsynaptic potential
Physiological Roles of NMDA Receptors in the CNS
- Wind up phenomenon
- Wind up = repeated stimuli of same strength cause increase in pain response
- Proposed mechanism: C fibres synapse in lamina II of dorsal horn → repeated stimulation → glutamate release → NMDA activation → ↑response of dorsal horn neurons to excitatory neurotransmitter input àhyperalgesia and allodynia
- Long term potentiation
- LTP = strengthening of synaptic transmission that occurs following ↑activity across that synapse
- i.e. recurrent painful stimuli → neuroplasticity → chronic pain
- NMDA receptor stimulation leads to various intracellular changes:
- production of NO
- activate second messengers (IP3, DAG, cGMP, PKC)
- PKC → increases NMDA activity → vicious cycle
- second messengers induce oncogenes (e.g. c-fos)
- Memory
- NMDA receptors are highly expressed in hippocampus and throughout cortex Important roles in neuroplasticity and memory formation
- Apoptosis
- Post cerebrovascular accident → significant NMDA activation → ↑↑Ca influx → excitotoxicity → triggers neuronal apoptosis
CICMWrecks Answer: Ketamine Pharmacology
Pharmacology of Ketamine
Examiner Comments
2022A 11: 74% of candidates passed this question.
The first part of this question required a description of both the receptor structure and its function. This includes, but is not limited to, its location, the natural ligand, how the channel may be regulated and the results of receptor stimulation. The second part of this question related to ketamine. Marks lost here often related to vague statements and incorrect facts. The examiners also commented that some candidates got confused between the R and S enantiomers. Few candidates commented on the nature of the metabolites and generally the PD section was vaguely answered.
2019A 15: 49% of candidates passed this question.
The NMDA receptor is a ligand gated voltage dependent ion channel located on post synaptic membranes throughout the CNS, with glutamate, an excitatory neurotransmitter, its natural ligand. A brief description of its structure, roles of glycine and magnesium, ions conducted, result of activation, role in memory and learning and agonists/antagonists was expected. Detail on structure and functions of the receptor were a common omission.
Ketamine, a phencyclidine derivative, is a non-competitive antagonist at the NMDA receptor. It is presented as a racemic mixture or as the single S(+) enantiomer (2-3 X potency).
Administration routes and doses scored marks. Pharmacodynamics were generally well covered including CVS (direct and indirect effects), CNS (anaesthesia, analgesia, amnesia, delirium, effects on CBF and ICP) respiratory (bronchodilator with preservation of airway reflexes) GIT effects (salivation, N and V). Knowledge of specific pharmacokinetic parameters was less well covered, including low oral bioavailability and protein binding and active metabolite (norketamine).
2010B 07
List the adverse properties of Propofol and Ketamine
CICMWrecks Answer
| PROPOFOL | KETAMINE | |
|---|---|---|
| Resp | – Respiratory depression – Apnoea – Decreased RR, TV, MV, FRC | – Mild respiratory stimulation – Respiratory depression if rapid IV administration – Increased secretions – Maintenance of pharygo-laryngeal reflexes > Laryngospasm – Bronchodilation > Likely Ca channel mediated via R-ketamine isomer |
| CVS | – Peripheral vasodilation – Bradycardia – Hypotension | – Indirect sympathomimetic effects → elevation of MAP > Due to inhibition of noradrenaline reuptake – Direct cardiodepressant effects |
| CNS | – Sedation – Decreased cerebral blood flow | – Increased cerebral blood flow – Increased cerebral O2 consumption – Sedation – Hallucinations – Emergence phenomena |
| GIT | – Nausea (although usually anti-emetic) | – Nausea > Due to serotonin reuptake inhibition |
| Other | – Propofol infusion syndrome > Rhabdomyolysis > Metabolic acidosis > Steatohepatitis – Injection site pain – Anaphylaxis – Leukocytosis | – Anaphylaxis – Increased muscle tone |
Examiner Comments
2010B 07: 6 (30%) of candidates passed this question.
This question was not well answered by many candidates. Candidates included advantages of these sedative / analgesic agents in their answers when they were not asked for in the question. The best approach to answer this question is to use a table listing their potential adverse properties in categories such as pharmaceutical and chemical properties, pharmacodynamic properties in different body systems, and pharmacokinetics. The common weaknesses observed included the side effects of ketamine, including its side effects on intracranial pressure, myocardial contractility and oxygen consumption, and hallucinations or delirium. Some adverse properties of propofol including bacterial contamination and pain on injection were also not well covered by many candidates. Syllabus: G2a,2a References: Goodman and Gilman The pharmacological basis of therapeutics 11th edition p350- 352
2007B 21
Compare and contrast the cardiovascular effects of an induction dose of Propofol and ketamine
CICMWrecks Answer
| PROPOFOL | KETAMINE | |
|---|---|---|
| Dose for induction | 2-2.5mg/kg 0.5-1mg/kg in critically unwell | 0.5-1.5mg/kg |
| Autonomic nervous system | Reduction in sympathetic outflow | Increase in sympathetic outflow Centrally mediated |
| Heart rate | Blunts baroreceptor reflex → no reflex tachycardia Bradycardia due to reduced sympathetic tone | Increased due to increased sympathetic outflow |
| Contractility | Mild direct negative inotropy Secondary effect due to reduced sympathetic tone | Direct negative inotropy Increased sympathetic tone → increased overall inotropy (providing maximal sympathetic tone has not already been reached by the patient) |
| Peripheral vascular resistance | Direct vasodilation Secondary effect due to reduced sympathetic outflow | Direct vasodilation Overall, increased due to increased sympathetic tone |
| Blood pressure | Decreased | Usually increased |
| Cerebral circulation | Reduced CMRO2 Reduced cerebral blood flow | Increased CMRO2 Increased cerebral blood flow (No way are we diving in to the ICP question in this debate!) |
Examiner Comments
2007B 21: 2 candidates (29%) passed this question
The key words in this questions were “compare and contrast”, “cardiovascular effects” and “induction dose”. Some candidates described aspects of both drugs other than the cardiovascular effects but gained no marks for this. Better answers used a combination of a table plus some explanation to contrast the cardiovascular effects of the two drugs concentration on aspects such as heart rate, cardiac output, vascular resistance and blood pressure.
Many candidates were confused by the direct versus the indirect cardiovascular effects of both drugs
Propofol probably has no direct negative inotropic effect. Ketamine has a direct myocardial depressant action but this effect is overridden by the centrally mediated sympathetic action of the drug. The effect of both drugs on the baroreceptor response alone is a difficult area as there is significant interplay between the direct cardiovascular effects of the drugs and their effect on the baroreceptor reflex. Allowance was made for this in the marking. Propofol resets the baroreceptor reflex producing a slower heart rate for a given level of blood pressure. Overall both drugs depress the baroreceptor reflex.
Comparison of the effects on cerebral, coronary, renal and hepatic blood flow earned extra marks.
2018B 04
Compare and contrast ketamine and midazolam.
Examiner Comments
2018B 04: 62% of candidates passed this question.
In addition to the key PK and PD properties of each drug, a clear comparison was required to score well (why choose one drug over the other?). When a table was used the addition of a comparison column was helpful.
A good answer covered the following: ketamine has analgesic properties whilst midazolam does not; ketamine preserves airway reflexes and does not cause respiratory depression unlike midazolam; whilst ketamine increases cerebral blood flow and CMRO2, midazolam decreases it; ketamine has a direct myocardial depressant effect which is often offset by an increase in sympathetic tone, whilst midazolam has no direct cardiac depressant effects but may reduce BP due to reduced SVR; midazolam has anticonvulsant properties, ketamine does not; ketamine is a bronchodilator; both drug effects are offset by redistribution; midazolam is lipophillic at body pH and will accumulate with prolonged infusions, ketamine will not; both are metabolised in the liver; midazolam can be reliably reversed by flumazenil, whereas there is no reliable complete reversal of ketamine; midazolam exhibits tolerance, dependence and withdrawal, whereas patients will only experience tolerance to the analgesic properties of ketamine.
“Drugs in Anaesthesia and Intensive care” chapters on midazolam and ketamine outline the key facts to include in this answer; interpretation and comparison of these facts will help achieve a good mark.\
2015A 22
Compare and contrast dexmedetomidine and ketamine
Examiner Comments
2015A 22: 50 % of candidates passed this question.
The majority of candidates were able to describe the mechanism of action, uses, dose and some side effects of each drug. The better answers were in a table format. It is of course possible to include much of the relevant information without using a table; however without the visual prompt of a table it makes it likely sections will be omitted.
When comparing two drugs it would be useful to note that though they both provide sedation with analgesia they are used in different circumstances. In ICU, dexmedetomidine is mainly used for sedation peri-extubation and may be continued post-extubation but this was not often mentioned.
The pharmacodynamic effects often omitted the cardiovascular and respiratory effects of ketamine (particularly bronchodilation).
The pharmacokinetic information required was not detailed but only minimal marks can be awarded for ‘administered IV with 100% bioavailability, liver metabolism and renal excretion’ which was a common answer. Noting dexmedetomidine is metabolised to inactive metabolites and ketamine is metabolised to norketamine gained marks, specific pathways were not required. Both drugs are licenced for administration intravenously (and ketamine may be administered IM); however other routes of administration are emerging in clinical practice for both drugs.
ii. Understand the pharmacology of local anaesthetic drugs.
2014A 17
Classify local anaesthetic agents and give examples. (30% of marks)
Describe the pharmacology of lignocaine. (70% of marks)
CICMWrecks Answer: Local Anaesthetic Classification
Local Anaesthetics
- Local anaesthetic agents act via inhibition of voltage gated Na+ channels, by binding to the intracellular portion of the channel
- → Decreased Na influx
- → Inhibition of depolarization
- → Inhibition of action potential formation and propagation
- Local anaesthetics are formed by a hydrophobic and hydrophillic component, and are classified according to the linkage between the two, either amide or ester
- Amide Local Anaesthetics
- e.g. Lignocaine, ropivacaine, bupivacaine
- Metabolized by hepatic dealkylation by P450 enzymes
- Longer duration of action
- Ester Local Anaesthetics
- e.g. Procainamide, cocaine
- Metabolized by plasma esterases
- Shorter duration of action
Mooney / JC 2019
CICMWrecks Answer: Pharmacology of Lidocaine / Lignocaine
Pharmacology of Lignocaine
Examiner Comments
2014A 17: 71% of candidates passed this question.
The first part of this question was answered well by most candidates.
Generally, the second part of the question was poorly organised by many candidates, the consequence being that many opportunities for picking up marks were lost. A brief statement as to what lignocaine is, its presentations and dose, some facts about PD and PK followed by a few lines on toxicity (CC/CNS ratio) was mostly what was required. Only a few candidates mentioned lignocaine toxicity
2021B 02 – 2019A 01
Describe the pharmacology of lidocaine.
Examiner Comments
2021B 02: 71% of candidates passed this question.
The answers for this question were generally of a good standard. Lidocaine is a core drug in intensive care practice and thus a high level of detail was expected. This question was best structured using a standard pharmacology template (pharmaceutics, pharmacokinetics and pharmacodynamics). A small number of answers omitted any pharmaceutic elements. Another common error was the use of vague and imprecise statements. For example, many answers stated that the maximum dose (without adrenaline) is 3 mg/kg, without specifying that this is subcutaneous.
The concept of the ratio of the dose required to produce cardiovascular collapse to that required to induce seizures (CC/CNS ratio) was often mentioned. However, in many cases this was conveyed simply as an abbreviated statement without any additional explanation leaving the examiner unsure as to whether the candidate understood the concept (and thus unable to award any additional marks). In addition, many candidates confused the order of this ratio (incorrectly referring to it as a CNS/CC ratio of 7). Lastly, few answers made specific mention of the narrow therapeutic index and the associated implications for use in the ICU.
2019A 01: 16% of candidates passed this question.
Comprehensive answers included uses (including antiarrhythmic action and a role in analgesia), physical properties and preparations, pharmacodynamics and pharmacokinetics. Its mode of action should also have been described. Many candidates focussed on toxicity and its management but provided little information on pharmacodynamics and pharmacokinetics, commonly omitting factors which affect its systemic absorption. Other common omissions were the dose required for its local anaesthetic effect and for its antiarrhythmic effect.
2015B 07
Draw and label a cross section of the lumbar epidural space (50% of marks).
Describe the pharmacology of bupivacaine (50% of marks).
CICMWrecks Answer: Anatomy of LP
Anatomy:
- Position in sitting or lateral decubitus position
- Level:
- Any of the interspaces between between L2-L5
- L4/5 interspace: Tuffier’s Line: line between iliac crests
- (or) L3/4 interspace: Line joining PSISs (Posterior superior iliac spines)
- Discrepancy between identified and actual intervertebral space in 50% of cases
- Conus medullaris ends at L1 in about 94% of patients
Tissues and target for LP:
- In the subarachnoid space between the arachnoid mater and the pia mater.
- The tissues pierced are (in order):
- skin
- subcutaneous tissue
- supraspinal ligament,
- interspinal ligament,
- ligamentum flavum,
- dura mater,
- the arachnoid mater into the subarachnoid space.
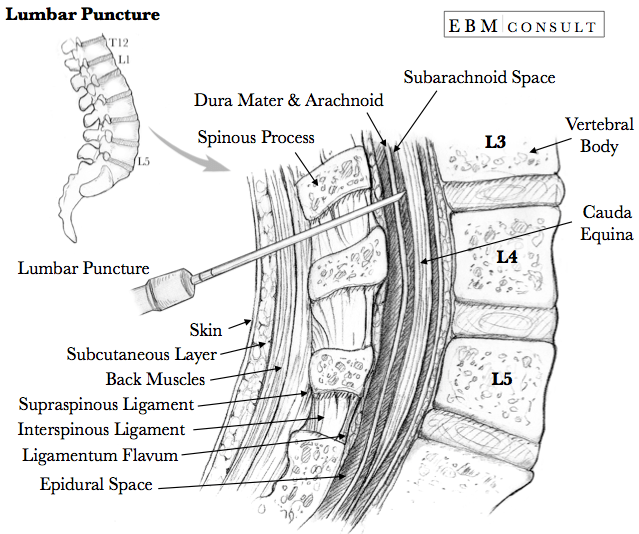
- Lateral/paraspinal approach: Skin – subcutaneous tissue – erector spinae muscles – ligamentum flavum – dura – arachnoid – subarachnoid space
Epidural Space
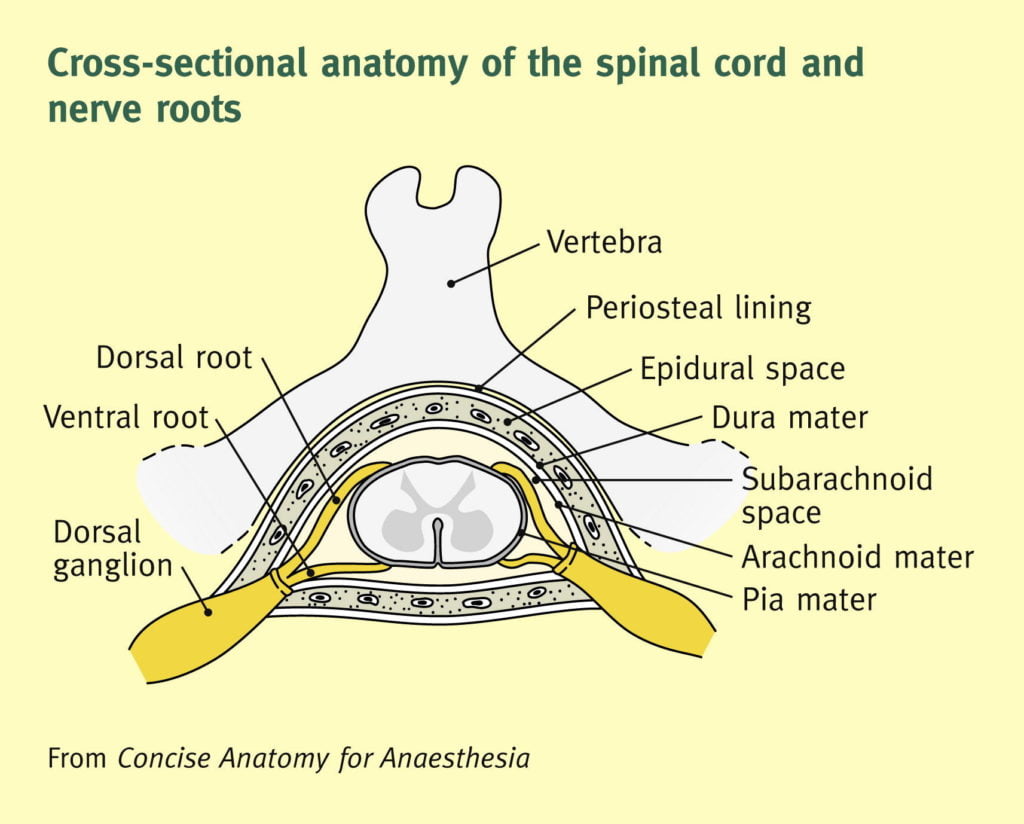
- Posterolateral Epidural Space
- Posterolateral epidural space extends vertically down the spinal canal and contains arteries, venous plexus, and fat
- Posterolateral epidural space is larger than the anterior epidural space
- Posterolateral epidural space is larger in the sacral region than it is in the cervical region
- Anterior Epidural Space
- Anterior epidural space is a virtual space under normal circumstances (due to adherence of dura to bone of vertebral bodies from the foramen magnum down to L1)
Gladwin / JC 2020
CICMWrecks Answer: Pharmacology of Bupivacaine
Examiner Comments
2015B 07: 35% of candidates passed this question.
It was expected answers would include a diagram of a cross section and label the lumbar epidural space and the key landmarks namely dura, subarachnoid space, epidural space. Most candidates were able to give a schematic representation even if not being able to draw. Some candidates confused the subdural space with the epidural space. Pharmacology of bupivacaine needed to cover both pharmacokinetics and pharmacodynamics. Several candidates addressed only one of these components and so missed the opportunity to score marks.
2009A 03
Outline the factors which affect the onset, duration of action and toxicity of local anaesthetic agents.
CICMWrecks Answer
Classification of Local Anaesthetics
- Esters: Procaine, Cocaine
- Unstable in solution
- Rapid hydrolysis by plasma cholinesterases
- ↑ freq of hypersensitivity reactions due to PABA byproduct
- Amides: Lignocaine, Prilocaine, Bupivacaine, Ropivacaine
- Stable in solution
- Slower hepatic metabolism
- Low rate of hypersensitivity reactions
Factors
| Factors affecting the… | Patient Factors | Drug Factors |
|---|---|---|
| Onset | 1. Tissue pH · infected (acidic tissue) → ↑ ionised portion → ↓ ΔC → slower onset 2. Nerve diameter · ↑ diameter → ↑ surface area → faster onset 3. Nerve firing rate · ↑ firing rate → faster onset (remember mechanism of reaching internal H-gate) 4. Pregnancy · ↑ Progesterone → ↑’d sensitivity to LA | 1. Ionisation factors · PKa · Weak bases (7.6-8.9) · Only ionised fraction transfers · pH < pKa → ↑ ionisation → ↓lipid solubility → ↓ rate of onset · Higher pKa → more effective once inside axon · Alkalinisation · addition of NaHCO3 → ↑ pH of preparation · ↑ pH → ↑ unionised fraction → ↑ rate of onset 2. Lipid solubility · ↑ lipid sol → ↑ potency → ↓dose → ↓’d Δ C · ↓ rate of onset 3. MW · ↓’d MW → ↑ rate of diffusion = ↑ rate of onset 4. Diffusion distance · ↓T → ↑ rate of onset 5. Area of diffusion · ↑ A (eg large nerve axon) → ↑ rate of onset |
| Duration of Action | 1. Site of administration · ↑’d regional blood flow → ↓’d duration 2. Metabolism · Esters metabolised faster than amides → ↓’d duration | 1. Protein binding · ↑ protein binding → longer duration 2. Intrinsic vasodilator/constrictor activity · Ropivicain has intrinsic vasoconstrictor activity · ↑ vasoconstrictor activity → ↑ duration 3. Lipid Solubility · ↑ lipid solubility → ↑ potency → ↓ dose → ↓ ΔC → ↓’d diffusion rate · This ↑’s sequestration into lipid rich compartments → ↑’d duration 4. Presence of Additives · Adrenalin → vasoconstriction → ↓’d systemic absorption → ↑’d ↑duration 5. Dose · ↑ dose → ↑ duration 6. Clearance · ↓ metabolism → ↑ duration · Amides slower metabolised that esters |
| Toxicity | Site of injection ↑tox with ◦ ↑local perfusion/Cardiac output ◦ low plasma cholinesterase activity (esters) ◦ hepatic or renal failure ◦ pregnancy | CC:CNS rato (higher is safer) – Lignocaine 7, ropivacaine 4, bupivacaine 3 ↑tox with ◦ ↑Dose ◦ Type (Amide>Ester) ◦ ↑Protein binding in tissues decreases systemic absorption, ↑protein binding in CNS = ↑toxicity ◦ ↑Na channel affinity (Bupivocaine slower to dissociate than lignocaine) ◦ ↑Conc – higher conc gradient between tissue and plasma, increasing systemic absorption ◦ ↓vasoconst additives ◦ pKa – LA’s are weak bases with high pKa’s; lower pKa = more unionized = ↑systemic absorption. Lignocaine pKa 7.9 (25% unionized at pH 7.4), bupivacaine pKa 8.4, 11% unionized at 7.4 |
Note: CC/CNS ratio was required (the ratio of plasma levels at which CVS Collapse vs. Convulsions occur).
Gladwin 2016
Examiner Comments
2009A 03: Pass rate: 10%
Marks were equally divided between all three parts. Structure to the answer using a table and list of facts gained credit. Factors affecting onset would be well described by stating Ficks law of diffusion and followed with an explanation of the equation. Factors affecting duration such as protein binding, regional blood flow, metabolism and use of vasoconstrictors scored marks.
Regarding toxicity, an explanation of the CC/CNS ratio was required (the ratio of plasma levels at which CVSCollapse vs. Convulsions occur). Other factors included structure of agents, accumulation e.g. due to liver disease. A mention of features of particular agents’ toxicity such as prilocaine and methaemoglobinaemia was expected.
Syllabus G2b 2a-c
References: Peck, Hill and Williams 2nd edition p163-174
Stoelting and Hillier 4th edition p179-203
Evers and Maze p507-533
2018B 19
Describe toxicity of local anaesthetic agents.
CICMWrecks Answer
Background
- toxicity = unwanted harmful effect of a drug
- LA= drugs that reversibly inhibit transmission of neural impulses in the applied region without affecting consciousness
- Toxicity from LA can be classified into local or systemic
- Local toxicity: mechanism of delivery + reaction to PABA
- Systemic toxicity = due to excess plasma concentration determined by rate of drug entrance relative to its redistribution and clearance by metabolism
Maximum recommended doses:
| Dose in mg/kg | Toxic plasma levels | CC:CNS ratio | |
|---|---|---|---|
| Lignocaine | 3mg/kg without adrenaline 7mg/kg with adrenaline | 5microg/ml | 7 |
| Bupivacaine | 2mg/kg up to 140mg +/- adrenaline | >1.5microg/ml | 3 |
| Ropivacaine | 3mg/kg +/- adrenaline | >4microg/ml | 5 |
Toxicity Mechanisms and Effects
Local toxicity
- High epidural or spinal blockade
- β fibres = small myelinated fibres readily blocked by LAs
- large doses of epidural/spinal LA can result in inappropriately high block
- High SY chain block
- Vaso + veno dilation → ↓SVR + ↑venous capacitance → ↓↓MAP
- Venodilation → ↓VR → ↓RAP → ↓HR
- Blockade of cardioaccelerator (T1-T4) fibres → ↓↓HR
- o Brainstem block
- Blockade of resp centre → profound resp depression
- Blockade of autonomic centre → CVS collapse (↓↓MAP + ↓↓HR)
- Neurotoxicity
- epidural/ intrathecal injection → neurotoxicity: mechanism: lignocaine ↑intracellular [Ca2+] → neurotoxic via unknown mechanism
- radicular irritation → transient neurological symptoms that resolve over 1-7days
- cauda equina syndrome → diffuse lumbosacral plexus injury → sensory, bladder + bowel dysfunction, paraparesis
- anterior spinal artery syndrome → lower limb paresis + dysaesthesia ?2o
anterior spinal artery spasm
Systemic toxicity
- CNS toxicity
- Biphasic effect
- Excitatory phenomena: depression of inhibitory interneurons → circumoral tingling, tongue numbness → restlessness, tinnitus, vertigo → skeletal + facial muscle twitching → seizure
- CNS depression: depression of central neurons → coma, apnoea
- Seizure ?2o inhibition of inhibitory GABAergic pathways → unopposed excitatory activities → seizure
- Biphasic effect
- CVS toxicity
- ↑↑dose required to produce CVS toxicity than CNS toxicity
- Main effects: hypotension + bradyarrhythmias
- Mechanism:
- Cardiac Na channel blockade LAs → slow conduction of cardiac impulses → PR prolongation + QRS widening → VTs
- Inhibit Ca2+ and K+ ion channels
- Inhibit cAMP synthesis (minor)
- Relaxation of arteriolar vascular smooth muscle
- CC:CNS ratio: ratio of the dose required to cause CVS collapse and the dose required to cause CNS toxicity: indicates that CNS is ↑vulnerable to LA than CVS
- Hypersensitivity reaction
- Rare
- LA or additive can → hypersensitivity reaction
- Ester LAs metabolised to para-aminobenzoate (↑rates allergy)
- Hepatotoxicity
- Continuous epidural infusion of bupivacaine found to rarely cause hepatic enzyme derangement + hepatoxicity
- Mechanism unclear
- Methaemoglobinaemia
- Prilocaine metabolised in liver to o-toluidine → oxidises Fe2+ of haem to Fe3+ → ↑methaemoglobin (unable to carry O2) → tissue hypoxia
Factors affecting risk of toxicity
- Drug factors
- Dose
- Route + location of administration
- Potency; pKa
- Isomerism (R-enantiomer more toxic than S)
- Rate of metabolism
- PB (↑PB → ↓free drug → ↓toxicity)
- Intrinsic vasodilator activity
- Patient factors
- Acidosis → ↑ionised → ion trapping → ↑toxicity
- Pregnancy → ↑progesterone → competitive binding to AAG → ↑unbound LA → ↑toxicity
- Hepatic/renal dysfunction
- Heart failure → ↓perfusion + ↓VD → ↓elimination
- Plasma cholinesterase activity
Prevention of LA toxicity
- Frequent aspirations
- Slow injection
- Test dose
- Total dose administered < max recommended dose
- Awake patient
- Monitoring
- USS guided
- Vasoconstrictors
Kerr 2016
Examiner Comments
2018B 19: 28% of candidates passed this question.
Most questions lacked a systematic approach to the question and specific detail. The relationship between systemic toxicity (CNS and CVS) and plasma levels should be described.
Many candidates did not clearly state that CNS toxicity occurs at lower plasma levels that CVS toxicity. Factors that affect toxicity (e.g. drug factors, patient factors, interactions) needed to be elaborated with some detail. Patient factors such as age, pregnancy, acidosis, hyperkalaemia, hepatic failure were often omitted. Finally, marks were also awarded for noting methaemoglobinaemia as possible toxicity and the existence of specific therapy (intralipid).
2012B 22
Describe the factors that increase the risk of systemic toxicity of the amide local anaesthetics
CICMWrecks Answer
Local anaesthetic
- Lipophilic group attached to ionized group via ester linkage (ester local anaesthetic) or amide group (amide local anaesthetic agents
- MoA
- Binds to and blocks volatage-gated Na channel on the inner surface of the cell membrane, inhibiting depolarization
- Increases threshold for depolarization
- Decreases impulse conduction
- Action potential abolished
- Higher affinity for open or inactive Na channels
- Hypercalcaemia stabilizes membrane (Na channels in rested state) reducing efficacy of local anaesthetics
- Hyperkalaemia depolarizes membrane (Na channels in open or inactive state) increasing the efficacty of local anaesthetics
Structure-Activity Relationship
- Ficks law of diffusion
- Membrane permeation
- Lipophilicity
- Small molecular weight
- Protein binding
- Can be displaced
- Membrane permeation
Toxicity
- CNS
- Paraesthesia in lips and tongue
- Convulsions
- Sedation
- Coma
- CVS
- Arrhythmia
Factors affecting local anaesthetic toxicity
Pharmacokinetics
A
- Dose
- Lignocaine 3~5mg/kg without adrenaline
- Ropivacaine 3mg/kg
- Bupivacaine 3mg/kg
- Route of administration
- IV
- Highly vascularized tissues
- Use of adrenaline decreases toxicity risk
D
- Lipophilicity
- pKa – Most LAs 7.9~8.4
- Low fraction of unionized drug
- pKa – Most LAs 7.9~8.4
- Size of drug
- Protein binding
- If high, less likely to cross BBB
- However if reaches cardiac myocytes, more likely to bind → prolonged effects
M and E
- Amide anaesthetic longer duration. Hepatic vs. ester metabolism
- Both renally excreted
Patient factors
- pH – will affect ionization state with acidosis reducing LA permeation, however once across the membrane, increased ionization increases LA binding to channel and effect
- Hepatic function
- Decreased metabolism à prolonged effect
- Decreased pasma protein à increased free fraction → increased effect
- Renal function
- Decreased GFR à decreased clearance → prolonged effect
Sakurai 2016
Examiner Comments
2012B 22: 4 (18.2%) of candidates passed.
The amide group of local anaesthetics consist of lignocaine, prilocaine, ropivacaine and bupivacaine. The systemic toxicity primarily relates to toxic plasma levels and the factors that influence this. The main factors expected can be categorized under drug factors (including kinetics), patient factors, site of injection and external factors. Many candidates omitted important details such as pKA, lipid solubility and addition of vasoconstrictors. For example, absorption is affected by drug pKA, (thecloser to physiological pH the more rapid the absorption), use of vasoconstrictors and the drugs own vasoactive properties, site of injection (intercostal>epidural>brachial plexus>subcutaneous infiltration). Distribution is dependent on physicochemical properties of the amide. The rate of metabolism, mechanism of action (bupivacaine, in comparison to lignocaine has stronger binding to inactivated resting sodium channels and a slower rate of dissociation) and external factors (e.g. systemic acidosis) are other factors that should have been mentioned, and expanded upon with relevant detail.
iii. Understand the pharmacology of anti-convulsant drugs.
2023B 04
Classify the mechanisms of action of anti-convulsant drugs (30% marks).
Outline the pharmacology of gabapentin (70% marks).
2008A 09
Describe the physiological basis for the mechanism of action of three commonly used anticonvulsant groups. Give an example of a drug for each mechanism of action.
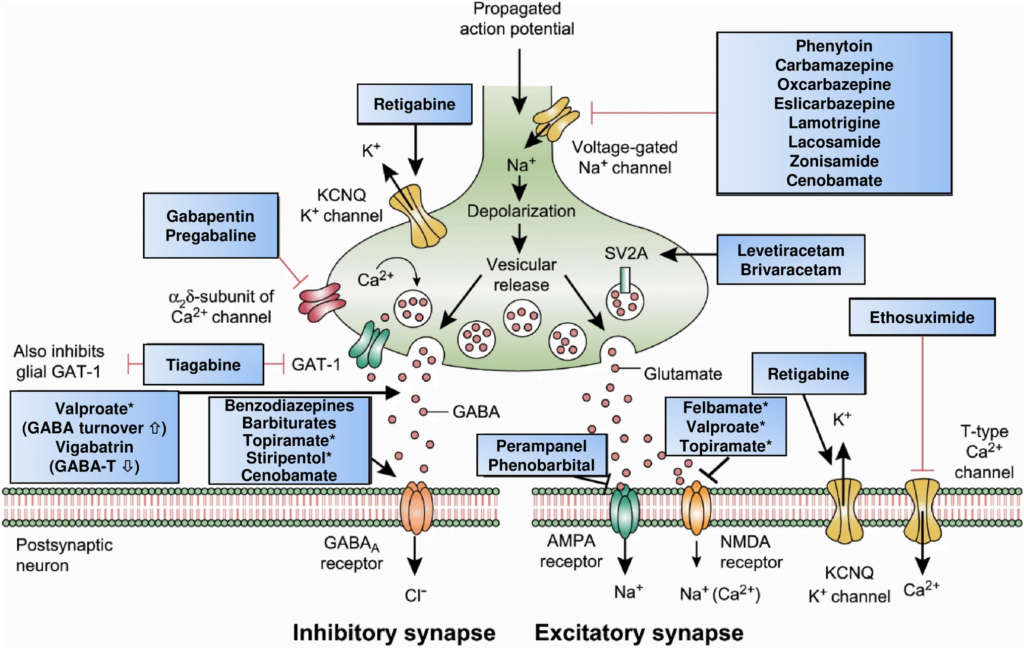
CICMWrecks Answer: Classification of Anti-Convulsants
Anticonvulsant Classification
(based on mechanism of action)
- Sodium channel blockers.
- Examples: phenytoin, carbamazepine, lamotrgine, Na valproate
- These promote the inactive state of voltage activated Na channels.
- Increased neuron refractory period to action potential generation.
- Rapid repetitive firing is diminished, spread of electrical activity to adjacent brain areas is suppressed.
- Drugs that enhance GABA mediated synaptic inhibition.
- Increases the influx of chloride ions into the cell and hyperpolarizes the neuron.
3 mechanisms:- Act on GABA receptor. Example: benzodiazepines, barbiturates.
- Inhibit GABA transporter and reduce neuronal GABA reuptake. Example: tiagabine.
- Promote GABA release. Example: gabapentin.
- Increases the influx of chloride ions into the cell and hyperpolarizes the neuron.
- Drugs that inhibit pre-synaptic Calcium channels.
- Example.: Na valproate, ?levetiracetam
- Limit activation of voltage activated Ca channel known as the T current
- Drugs that inhibit AMPA Kinate.
- Example.: Topiramate
- Limit proexcitatory action of glutamate
- Drugs that NT release.
- Example.: ?levetiracetam
- Binds to synaptic vesicle glycoprotein SV2A reducing exocytosis of synaptic vesicles
CICMWrecks Answer: Pharmacology of Gabapentin
Examiner Comments
2023B 04: 21% of candidates passed this question.
The first part of this question required candidates to correlate the mechanism of action of anti epileptic drugs with atleast one example under each section. These included GABA potentiation (Barbiturates/Tiaganbine), reduction in excitory transmission (including NMDA/AMPA antagonists) and modification of ionic conductances (sodium channel blockade-Phenytoin, Calcium channel blockadeLamotrigine, Gabapentin, activation of potassium channels- Retigabine).
The second part of this question involved an overview of gabapentin pharmacology. Gabapentin is an amino acid analogue of GABA utilised in neuropathic pain and epilepsy. There a multiple proposed mechanisms of action including Ca+ antagonism, inhibited glutamate release and anatagonism and increased GABA concentrations.. Important pharmacokinetic properties included dose dependent absorption, minimal protein binding, no metabolism with renal clearance and requirement for dose reduction in renal failure. It has largely central neurological side effects with a rare but important association with steven johnson syndrome or toxic epidermal necrolysis.
2008A 09: 1 candidate (33%) passed this question.
The three main anticonvulsant mechanisms required were:
- Sodium channel blockers. These promote the inactive state of voltage activated Na channels. Sodium channels are unable to open for a period of time making the neuron more refractory to action potential generation. Rapid repetitive firing is diminished and spread of electrical activity to adjacent brain areas is suppressed.
Examples: phenytoin, carbamazepine, lamotrgine, Na valproate - Drugs that enhance GABA mediated synaptic inhibition. This increases the influx of chloride ions into the cell and hyperpolarizes the neuron. 3 mechanisms:
a. Act on GABA receptor. Example: benzodiazepines, barbiturates.
b. Inhibit GABA transporter and reduce neuronal GABA reuptake. Example: tiagabine.
c. Promote GABA release. Example: gabapentin. - Drugs that inhibit Calcium channels. Limit activation of voltage activated Ca channel known as the T current. Example.: Na valproate
Other mechanisms of action with examples if described earned extra marks. These included glutamate /NMDA receptor inhibition. Example: magnesium.
Syllabus G2f
2012A 03 – 2010A 02
Describe the pharmacology of phenytoin
Examiner Comments
2012A 03: 6 (60%) of candidates passed.
Many candidates scored well in this question by using a standardised approach such as: Pharmacoceutics, Pharmacokinetics and Pharmacodynamics. This topic is well covered in the reference texts and a high degree of content was expected.
2010A 02: 9 (90%) of candidates passed this question
A structured approach was expected addressing both the mechanism of action and pharmacokinetics. Candidates were expected to outline relevant mechanisms of action (such as sodium channel blockade) and how they relate to its use as an anticonvulsant agent. Additional credit was given for discussing other potential mechanisms and other uses such as pain management and antiarrhythmic properties.
Phenytoin is illustrative of several key concepts in pharmacology and mention of these was expected. Failure to address these key concepts or provide sufficient detail was a common omission. Candidates were expected to discuss that phenytoin is highly protein bound, changes from first to zero order kinetics with escalating doses and is metabolised by the cytochrome p450 enzyme system. Some discussion of the significance of these points was expected and extra credit was awarded for more detailed explanations, comments on enzyme induction and examples of drug interactions that are well known and clinically relevant. Candidates were expected to
comment on the mode of delivery and compare oral and intravenous dosing. It was expected that the need for a loading dose followed by maintenance dosing would be mentioned and extra credit was given for highlighting the potential hazards of rapid intravenous administration. Additional credit was given for mentioning the importance of a narrow therapeutic index and the need for clinical monitoring. Well organized answers such as those with an ordered list of subheadings were rewarded.
Syllabus: G2f, 2f
References: Goodman and Gilman’s the Pharmacological Basis of Therapeutics,
Chp 19
2017A 17
Compare and contrast the pharmacology of phenytoin and levetiracetam.
2014B 02
Describe the pharmacology of Phenytoin (75% of marks) and Levetiracetam (25% of marks).
Examiner Comments
2017A 17: 35% of candidates passed this question.
A table was useful to answer this question. Comparing and contrasting the pharmacology was required to score well rather than listing various aspects of pharmacology. The key properties of the drugs which demonstrate their importance to ICU was required.
2014B 02: 35% of candidates passed this question.
The knowledge of phenytoin was often superficial and many answers were too brief and didn’t adequately cover the required material. The knowledge around levetiracetam seemed very limited with many candidates guessing (incorrectly) what the pharmacokinetics might be. Most answers demonstrated a structured approach to this type of question.
Better answers were able to distil major issues such as the narrow therapeutic window for phenytoin or the potential clinical impact of differing ordered kinetics or altered metabolism. Candidates are reminded to read each question carefully; levetiracetam should not be confused with levosimendan.
2015B 24
Compare and contrast the pharmacology of valproic acid and carbamazepine
Examiner Comments
2015B 24: 6% of candidates passed this question.
Both these agents are listed as “level B” in the syllabus pharmacopeia and as such a general understanding of each class and relevant pharmacokinetics and pharmacodynamics was expected. Most candidates had better knowledge of valproate than carbamazepine. Some description of the toxicological features for intensive care practitioners was expected.
iv. Understand the pharmacology of anti-depressant and anti-psychotic drugs.
2009A 21
Compare and contrast the mechanism of action and side effects of tricyclic antidepressants, selective serotonin reuptake inhibitors and monoamine oxidase inhibitors.
CICMWrecks Answer
| TRICYCLIC | SSRI | SNRI (Not in this qn) | MAOI | |
|---|---|---|---|---|
| E.g. | Amitriptyline | Fluoxetine | Venlafaxine | Phenylzine |
| PK | Well-absorbed PO with high first pass. Highly protein bound. Very lipid soluble. Hepatic phase 1 and 2 metabolism Renal excretion T1/2= 30-45 hours | 50:50 racemic mixture Well absorbed Hepatic metabolism Does require dose adjustment in renal failure for metabolites | High first pass- F=45% Large Vd with much lower PB (30%) Hepatic 2D6 metabolism to desvenlafaxine (active) Renal elimination. T1/2= 10 hours | Incomplete data available Renal elimination of metabolites Duration depends on time taken for MAO synthesis |
| MoA | Competitively inhibit neuronal uptake (uptake 1) of noradrenaline and serotonin, thereby increasing their concentrations in the synapse Additional: 1. Antimuscarinic 2. Antihistamine 3. Alpha-adrenoreceptor antagonism | Inhibition of SERT transporter increases synaptic 5-HT. No effect on NET | Inhibition of serotonin re-uptake (SERT) and noradrenaline re-uptake (NET). Low affinity for NET at low doses. Little muscarinic, H1 or alpha1 antagonism effects | Irreversible MAO A and B inhibition leading to accumulation of: 1. Noradrenaline 2. Serotonin 3. Dopamine |
| A/E | 1. Initially increase suicidality in youth 2. CNS- sedation (H1), seizures 3. Anti-Ach: dry mouth, constipation, urinary retention, blurred vision 4. CVS- postural hypotension | Serotonin A/E (shared by all AD): – Insomnia/somnolence – N/V/D – Sexual- low libido, impotence – Initial suicidality | 1. Serotonin A/E 2. Norad- HTN, tachycardia | 1. Serotonin 2. Histamine 3. Anticholinergic 4. Anti-alpha 5. Adrenergic |
| OD | 1. CVS – Sinus tachy – QT prolongation – QRS prolongation → VT/VF (Sodium channel blockade) – RBBB – Labile BP 2. CNS – Excitation and seizures then depression 3. Mydriasis, hyperthermia Treatment: 1. Benzos for seizures 2. Hyperventilation and sodium bicarbonate if QRS prolonged 3. Avoid inotropes if possible | 1. Serotonin Syndrome | 1. Serotonin syndrome 2. Cardiotoxic | 1. Serotonin syndrome Interacts with other antidepressants, foods high in tyramine (cheese) and sympathomimetic |
Ruan 2020
Examiner Comments
2009A 21: Pass rate: 40%
Good answers were in tabular format.
The antidepressant action is similar for each agent. Initial increase in 5HT and NA, followed in 2-3 weeks by a down regulation or change in efficiency of 5HT transmission. The agents produce elevated neurotransmitters via different mechanisms, either reuptake blockade or enzyme inhibition. MAOIs can be competitive or non-competitive. Mention of the different neurotransmitters affected by each agent was required.
A description of significant side effects at therapeutic doses, and in overdose was expected with explanations provided. These should have included – the anticholinergic effects and cardiotoxicity of TCAs, postural hypotension, the catecholamine, pethidine and tyramine related complications of MAOIs, and serotonin syndrome with SSRI/MAOI use and or overdose. More marks were gained for mention that side effect profiles can be beneficial e.g. analgesic properties of TCAs, sedation with TCAs/ SSRIs and energizing benefits of SSRIs/SNRIs. SSRI’s safety and efficacy have markedly reduced the use of MAOIs and to a lesser extent TCA’s..
Syllabus G2f2d
Reference: Stoelting p 398-407, Katzung p 476-487.
2010A 13
Describe the physiological basis of the effects seen in the serotonin syndrome (80% marks). List the classes of drugs that may cause the serotonin syndrome (20% marks).
CICMWrecks Answer
Serotonin (5-HT or 5-Hydroxytryptophan)
- an amine neurotransmitter mainly in the CNS and GI tract
- Action on Family of receptors
- Gq-Coupled, Excitatory, ↑ DAG and IP3
- 5-HT2: CNS/PNS, GIT, Plts and Blood vessels
- Gs-Coupled, Excitatory, ↑ cAMP
- 5-HT4: CNS/PNS, GIT
- 5-HT6: CNS
- 5-HT7: CNS,GIT and Blood vessels
- Gi-Coupled, Inhibitory, ↓ cAMP
- 5-HT1: CNS and Blood Vessels
- 5-HT5: CNS
- Ligand gated Na or K, Excitatory
- 5-HT3: CNS/PNS and GIT
- Gq-Coupled, Excitatory, ↑ DAG and IP3
- Main actions
- CNS
- Inhibitory effect on pain pathways
- Inhibitory effect on higher cortical function, stabilising mood and decreasing wakefulness
- CVS:
- 5HT-2 – SMC, vasoconstriction
- Direct small positive chronotropic and inotropic effects
- GIT:
- ↑↑ tone and peristalsis in the GI tract
- CNS
Symptoms of Serotonin Syndrome
- Increased levels of serotonin in the CNS
- CNS Mediated
- hyper-reflexia
- tremor
- clonus
- skeletal muscle contraction and hyperthermia
- Peripheral effects
- hypertension
- GIT effects
- diarrhoea.
Classes of Drugs which can cause Serotonin Syndrome
- Antidepressants
- SSRI, SNRI, MAOi, TCA
- Other
- amphetamines
- pethidine, tramadol
- Methylene Blue
- Sumatriptan (5-HT1 agonist)
Gladwin 2016
Examiner Comments
2010A 13: 7 (70%) of candidates passed this question.
For a good answer candidates were expected to mention the role of serotonin (5
hydroxytryptamine) is an important neurotransmitter, a local hormone in the GIT and
involved in platelet reactions. It is formed from tryptophan and metabolised by MAO
(thus the potential effect of a combination SSRI with MAO inhibitor, or concurrent
use of several serotonin affecting drugs). Typically the serotonin syndrome is a
predictable effect of increased CNS levels of serotonin with a consequence of hyper
reflexia, tremor, clonus, skeletal muscle contraction and hyperthermia (but in
serotonin syndrome these effects are probably CNS mediated), hypertension, and
diarrhoea.
The expected list of drugs included the SSRI’s themselves, combination of SSRI’s
and MAOI’s, antidepressants (2nd generation [e.g Venlafaxine]), Tramadol (blocks
serotonin re-uptake), pethidine, fentanyl, ondansetron, sumatriptan (5-HT1 agonist).
Syllabus: M3
References: Basic and Clinical Pharmacology, Katsung pg 264 – 269
2007B 03
Describe the physiological effects and principals of management of tricyclic antidepressant overdose
CICMWrecks Answer
TCA (e.g. amitryptilline and imipramine)
- Toxic dose of amitrytilline
- >10mg/kg
Mechanism of Action
- Act via central inhibition of noradrenaline reuptake (via inhibition of NET) and serotonin reuptake (via inhibition of SERT) but not dopamine transfer
- Also Na channel blockade
- Agonist at α1 and to a lesser extent α2 receptors
- Decreases sensitivity of muscarinic receptors
- Antagonist at H1 receptors
- Downregulates expression of GABAb and NMDA receptors
Physiological effects
CNS
- Agitation
- Seizures
- Coma and respiratory depression
CVS
- Sinus tachycardia
- Hypertension
- Na channel blockade
- Prolonged QT
- Widened QRS
- RBBB
Anticholinergic
- Dry mouth
- Dry, warm skin
- Urinary retention
- Tachycardia
Management
- Maintain airway patency
- Support ventilation
- Support organ perfusion
- Fluids, vasopressors as needed
- ABGs, blood tests, ECG
- Activated charcoal early
- However TCA rapidly absorbed
- Sodium bicarbonate
- 100mmol boluses
- Target pH >7.45
- Alkalinization promotes protein binding and limits free fraction of drug
- Alkalinization promotes dissociation of TCA and Na channel
- Consider intralipid, hypertonic saline for refractory cases
Examiner Comments
2007B 03: 1 candidate (14%) passed this question.
Good answers to this question were those that gave an accurate account of the physiological effects, e.g. inhibition of the fast sodium channels in the His-Purkinje system as well as the atrial and ventricular myocardium, decreasing conduction velocity (differential conduction inhibition of RBB being more susceptible) and increasing duration of repolarization, and the absolute refractory periods. Once having done that the rest of the answer would have flowed more easily, e.g. ECG changes and conduction disturbances. The effects of tricyclic antidepressants on Na channels and as a consequence the cardiovascular conduction abnormalities were often omitted. Anti cholinergic (e.g. slowing GIT motility) and antihistamine effects (e.g. obtundation) were often overlooked. Also frequently overlooked were basic pharmacology relevant to treatment, e.g. lipophilic, large volume of distribution, systemic acidosis reduces the extent of protein binding and increases unbound (active) drug. Additional points were available to those who not only mentioned sodium bicarbonate, but also mentioned the principles behind its use for this circumstance
2021B 16
Classify the anti-psychotic drugs (25% marks). Outline the pharmacology of haloperidol (75% marks).
CICMWrecks Answer: Classification of Anti-Psychotics
ANTI-PSYCHOTIC DRUGS
CLASSIFICATION
| TYPICAL Antipsychotic Agents | ATYPICAL Antipsychotic Agents | |
|---|---|---|
| (D2-R) | (D2-R<5-HT2a) | |
| Includes | (1) Phenothiazines (Eg. Chlorpromazine) (2) Butyrophenone (Eg. Haloperidol) (3) Thioxanthenes (Eg. Flupenthixole) | (1) Diazepines (Eg. Clozapine, Olanzapine) (2) Dibenzothiazepines (Eg. Quetiapine) (3) Benzamides (Eg. Sulpiride) (4) Benzisoxazols (Eg. Respirdone) |
| Mechanism of Action | (i) Primarily potent D2R antagonists (> 80% of receptors must be antagonised for therapeutic effect) (ii) Moderate antagonism of 5-HT2 receptors and α-adrenoceptor (iii) Weakly inhibit mAChR and H1R | (i) Highly selective D2R antagonists (> 80% of receptors must be antagonised for therapeutic effect) (ii) High affinity for 5-HT2R and moderate affinity for α-adrenoceptors (Except benzamides) (iii) Diazepines have D4R antagonistic effect also |
| Effect | Ameliorate +ve symptoms only (little effect on –ve symptoms) | Indicated for treatment resistance or side-effects with typical agents (esp extrapyramidal symptoms) Have greater therapeutic efficacy (ameliorate both +ve and –ve symptoms) |
| Side Effects | Very narrow TI – Substantial side-effects at therapeutic doses (i) Movement disorders: – Extrapyramidal symptoms, such as dystonias, akathisias, pseudoparkinsonism – These are acute and reversible – Tardive dyskinesia (involuntary movements of tongue, lips, face, trunk and extremities) – Gradual onset and irreversible (ii) Endocrine – Sexual dysfunction, gynaecomastia (due to PRL release), menstrual disorders, weight gain (iii) Hypotension (α-adrenoceptor-related) (iv) Sedation (H1R related) (v) Seizures (vi) Jaundice and agranulocytosis (esp with phenothiazines) (vii) Anticholinergic symptoms (viii) Neuroleptic malignant syndrome | fewer side effects than typical agents (esp movement disorders and gynaecomastia) (i) Cardiac arrhythmias (ii) seizures (iii) clozapine-induced agranulocytosis (requires blood screening) (iv) myocarditis and cardiomyopathy (requires cardiac enzyme and TTE monitoring) |
CICMWrecks Answer: Pharmacology of Haloperidol
PHARMACOLOGY OF HALOPERIDOL
Examiner Comments
2021B 16: 28% of candidates passed this question.
Excellent answers were able to provide a classification of antipsychotics based on either typical/atypical or first/second generation categories, provide examples of each and identify key differences in mechanism and effects. They also distinguished between butyrophenones and phenothiazines within the typical antipsychotic group. Haloperidol was identified as a butyrophenone, with description of pharmaceutics, dose and route, as well as pharmacodynamics and pharmacokinetics. Key adverse effects were detailed, focusing on those specific to haloperidol, including a description of different types of extrapyramidal symptoms and QT prolongation/ torsades de pointes.
2014B 21
Compare and contrast the pharmacology of haloperidol and diazepam.
Examiner Comments
2014B 21: 50% of candidates passed this question.
These are both commonly used agents and a tabulated format worked well. Subheadings covering the “general” pharmacology approach ensured core areas were addressed. Vague terms such as “good” or “moderate” did not allow a detailed comparison between the agents. Repetition of facts between sections such as uses, pharmacodynamics, effects and adverse effects did not gain further marks
Recent Comments