Syllabus (Fourth Edition, 2023)
Topics
i. Explain single and multiple compartment models.
ii. Describe the absorption of drugs and factors that influence this.
iii. Describe the distribution of drugs and factors that influence this.
iv. Describe the mechanisms of drug metabolism and clearance.
v. Explain the kinetics of an intravenous bolus and infusion.
vi. Describe the concepts of effect-site concentration and context sensitive half-time.
vii. Explain clinical drug monitoring with regard to peak and trough concentrations, minimum therapeutic concentration and toxicity.
viii. Describe the pharmacokinetics of drugs in the epidural and subarachnoid space.
Topics not covered in previous SAQs
i. Explain single and multiple compartment models.
v. Explain the kinetics of an intravenous bolus and infusion.
vi. Describe the concepts of effect-site concentration and context sensitive half-time.
vii. Explain clinical drug monitoring with regard to peak and trough concentrations, minimum therapeutic concentration and toxicity.
viii. Describe the pharmacokinetics of drugs in the epidural and subarachnoid space.
Learning Objectives for the First Part Examination in Intensive Care Medicine
- This will ensure that trainees, tutors, and examiners can work from a common base.
- All examination questions are based around this Syllabus.
- These learning objectives are designed to outline the minimum level of understanding required for each topic.
- The accompanying texts are recommended on the basis that the material contained within them provides sufficient information for trainees to meet the learning objectives.
- Trainees are strongly encouraged to explore the existing and evolving body of knowledge of the Basic Sciences as they apply to Intensive Care Medicine by reading widely.
- For all sections of the syllabus an understanding of normal physiology and physiology at extremes of age, obesity, pregnancy (including foetal) and disease (particularly critical illness) is expected.
- Similarly, for pharmacology, trainees are expected to understand a drug’s pharmacology in these contexts.
- An understanding of potential toxicity and relevant antidotes is also expected.
Definitions
Throughout the document specific wording has been used under the required abilities to indicate the level of knowledge and understanding expected and a glossary of these terms is provided.
Definitions
| Calculate | Work out or estimate using mathematical principles. |
| Classify | Divide into categories; organise, arrange. |
| Compare and contrast | Examine similarities and differences. |
| Define | Give the precise meaning. |
| Describe | Give a detailed account of. |
| Explain | Make plain. |
| Interpret | Explain the meaning or significance. |
| Outline | Provide a summary of the important points. |
| Relate | Show a connection between. |
| Understand | Appreciate the details of; comprehend. |
SAQs
i. Explain single and multiple compartment models.
ii. Describe the absorption of drugs and factors that influence this.
2020A 15 – 2017B 22
Define bioavailability. (10%) Outline the factors which affect it. (90%)
CICMWrecks Answer
DEFINITION
The fraction of drug that reaches the circulation compared with the same dose given intravenously. (%)
(or)
The ratio of the area under the stated concentration–time curve (AUC) divided by the area under the i.v. concentration–time curve. (%)
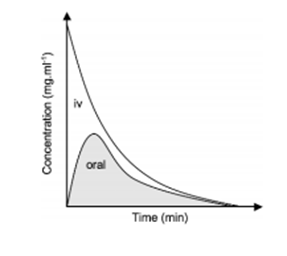
FACTORS AFFECTING BIOAVAILABILITY
- Pharmaceutical factors
- Preparation: Decreasing order: Solutions > Suspensions > Capsule > Tablet > Coated tablet
- Particle Size
- Salt form
- Crystal forms have better availability compared to amorphous forms
- Degree of ionization: – Non-ionised, lipid soluble drugs – higher bioavailability
- Pharmacological factors
- Gastric Emptying and GI Motility
- GI Diseases: Coeliac, Crohn’s
- Timing in relation to food intake
- First pass metabolism: The degree of metabolic breakdown of an orally administered drug that occurs in the intestine or liver before it reaches the systemic circulation.
- Drug-Drug interactions
- Pharmacogenetic factors
- Other factors:
- Area of absorptive surface
- State of circulation (shock, tissue perfusion)
- Hepatic Insufficiency, Poor renal function
- Route of administration
Sources: Cross and Plunkett. Physics, Pharmacology, Physiology for Anaesthetists. Goodman & Gilman’s Pharmacological Basis of Therapeutics. Katzung Clinical Pharmacology
JC 2020
Examiner Comments
2020A 15: 49% of candidates passed this question.
Many candidates spent time defining and describing aspects of pharmacokinetics which were not relevant to the question. E.g. clearance, volume of distribution and half-life. Candidates who scored well utilised a structure which incorporated the headings of the factors which affect the bioavailability of medications with a simple description as to the nature of the effect. These factors included: the physical properties of the drug, the preparation, patient factors, the route of administration and metabolism amongst others.
2017B 22: 33% of candidates passed this question.
Many candidates did not specify that bioavailability describes the proportion/fraction of drug reaching the systemic circulation (to differentiate from the portal circulation). Some candidates considered only factors impacting absorption from the GI tract or stated that bioavailability related only to orally administered drugs. Candidates failed to provide an equation, or got equations or graphs wrong. Nearly all candidates failed to provide a comprehensive list of factors affecting bioavailability.
2010A 17
With regard to ORAL drug dosing, describe the factors that affect the fraction of drug reaching the systemic circulation (80% marks).
How may these factors be altered in a patient with shock (20% marks)?
CICMWrecks Answer
Oral Bioavailability
Absorption
- Drug factors
- Most drugs absorbed from GI via passive diffusion
- Diffusion:
- Low molecular weight substances are readily able to permeate membranes (*see below for equation)
- Charge
- Charged particles display reduced permeability across phospholipid bilayers
- pKa → pH at which 50% of the molecules are ionized
- Henderson-Hasselbach Equation pKa = pH + log (ionized/unionized)
- Acids are ionized when pH > pKa
- Bases are ionized when pH < pKa
- Hydrophilicity
- A degree of hydrophilicity is required for drug to cross aqueous layer immediately adjacent to enterocyte in intestinal lumen
- Lipophilicity
- Lipophilicity required for drug to cross phospholipid bilayer
- Pharmaceutics
- Dissolution of tablet/capsule
- Enteric coating
- Diffusion:
- Most drugs absorbed from GI via passive diffusion
- Patient factors
- Rate of gastric emptying
- Absorption surface area greater in intestines (>200m2) due to microvilli, therefore increased gastric emptying increases rate of absorption
- Blood flow to site of absorption
- Reverse transporters such as P-glycoprotein cause drug efflux from enterocytes, preventing absorption
- Inflammation → increases drug absorption
- Interaction with other co-administered drugs
- Rate of gastric emptying
Fick’s Law:
First Pass Metabolism
- Metabolism by gut bacteria (digoxin)
- Metabolism within enterocytes
- Metabolism in liver via portal circulation before reaching systemic circulation
- Secretion into bile by liver on first pass, before reaching systemic circulation
Shock
Introduction
Clinical state of acute circulatory failure with impaired oxygen utilization +/- delivery by cells leading to cellular hypoxia or dysoxia
- Hypovolaemic (Massive haemorrhage)
- Obstructive (e.g. Massive PE, tamponade)
- Cardiogenic (Acute cardiac failure)
- Distributive (Sepsis)
Effects on Bioavailability
- Drug factors will be unaltered in a patient in shock
- Patient factors
- Gastric stasis may occur leading to decreased rate of absorption
- Intestinal barrier – the integrity of the GI barrier may be decreased by inflammation or ischaemia and cell death, leading to greater absorption
- Splanchnic blood flow may be decreased leading to decreased absorption
- P-glycoprotein may be inhibited due to cell dysfunction or drug interactions (e.g. antibiotics) leading to decreased efflux → increased drug absorption
- 1st pass metabolism
- Concurrent treatment with antibiotics may alter the microbiota in the GI lumen, altering metabolism
- Hepatic dysfunction may occur due to ischaemia or haemolysis
- If hypoxia → zone 3 hepatocytes will be relatively impaired → CYP450 metabolism decreased → increased bioavailability
Sakurai 2016
Examiner Comments
6 (60%) of candidates passed this question
For a good answer candidates were expected to define bioavailability and the factors that affected a drugs oral bioavailability. This was often overlooked by candidates. For example, factors affecting absorption (Metabolism by gut flora, drug / drug interactions with in the gut, lipophilicity and hydrophilicity of the drug (drug that are markedly lipophilic or hydrophilic cross the mucus layer or villous membrane poorly), First Pass clearance and sites and mechanism of possible metabolism, to define hepatic clearance, extraction ratio (providing a formula proved helpful to many candidates) and factors that affected hepatic drug clearance. Candidates often lacked an understanding of this area, or failed to mention it. In relation to the second part of the question, candidates were expected to mention the effects of reduced absorption and altered first pass metabolism resulting in uncertain bioavailability of oral drugs.
Syllabus: Section II, 2a, b References: Pharmacology, Rang, Ritter and Dale, Chp 7. Goodman and Gilman’s the Pharmacological Basis of Therapeutics, Chp 1
2023A 02 – 2014A 20
Describe the factors affecting drug absorption from the gastrointestinal tract.
CICMWrecks Answer
Drug Factors
- Lipid solubility
◦ ↑ lipid sol → ↑ absorption
◦ Must still have some water soluble component - Molecular size
◦ ↓ size → ↑ rate of absorption
◦ Larger molecules require processes other than simple diffusion - Degree of ionisation
◦ pH-pKa gives the degree of ionisation
◦ ↓’d ionisation favours absorption - Physical forms
◦ Gases faster than liquids faster than solids - Dosage forms
◦ Capsules more rapidly absorbed than tablets - Formulation
◦ The addition of Na to the formulation for stability can reduce the bioabalability via the oral route - Concentration
◦ From Fick
Patient factors
- Area of absorptive surface
◦ intestinal resection or coeliac → ↓ absorptive surface → ↓ absorption from Fic - Vascularity
◦ Shock → ↓ absorption - pH
◦ effect degree of ionisation
▪ ↓ pH favours absorption of acidic drugs
▪ ↑ pH favours absorption of basic drugs - Presence of other substances
◦ Statins should be taken with food
◦ Milk ↓’s and Vit C ↑’s absorption of iron - GI motility
◦ ↑ motility → ↓ absorption - Functional Integrity of the absorptive surface
◦ Coeliac disease → flattened mucosa → ↓ absorption - Coexisting disease state
◦ diarrhoea → ↓ absorption
Gladwin 2016
Examiner Comments
2023A 02: 39% of candidates passed this question.
This question required candidates to draw on knowledge across the syllabus. Many candidates did not appreciate the breadth required to adequately address this question unfortunately providing very detailed information on only aspects of the answer required to pass.
Excellent answers described the following: differences between different GI routes/ sites of absorption, mechanisms of absorption including Fick’s law of diffusion, drug formulations, drug factors (including concentration gradient, lipid solubility, ionisation, and molecular weight), site factors (including surface area, first pass metabolism, GI wall thickness, blood flow, and GI motility/ transit time). Marks were also given for consideration of drug interactions, enterohepatic recycling, and luminally active drugs.
2014A 20: 45% of candidates passed this question.
This is a very broad and open question. While a structured approach was useful, a sound knowledge of first principles or even being able to “think on the fly” would have provided candidates with enough opportunities to generate a pass.
iii. Describe the distribution of drugs and factors that influence this.
2019A 05
Define volume of distribution (15% of marks).
Outline the factors affecting volume of distribution (60% of marks)
and explain how it may be measured (25% of marks).
2015B 12
Define “volume of distribution” and describe the factors that influence it.
CICMWrecks Answer
Definition
Volume of Distribution (Vd) = The theoretical volume of bodily fluid in which a drug would need to be distributed following its administration to produce the observed / desired plasma concentration
Mathematically, at time t=0:
Factors Influencing Volume of Distribution
Drug factors
- ↑ Plasma protein binding
- ↓’s Vd
- ↑ Tissue binding
- Binds to tissues → ↓ circulating drug → apparent ↑ Vd
- Physicochemical properties of drug
- ↑ Lipophilicity → ↑ Vd
- ↑ Hydrophilicity → ↓Vd
- ↓ Size → ↑ Vd
- ↓ Charge → ↑ Vd
- pKa
- Basic drug → lower pKa = ↑ unionised portion → ↑ Vd
- Acidic drug → higher pKa = ↑ unionised portion → ↑ Vd
Patient factors
- ↓ pH → ↑’d ionisation of basic drugs → ↓ diffusion → ↓ Vd
- Blood flow
- ↑ blood flow → ↓d concentration → ↑ Vd
- Age
- ↑ Vd in neonates
- Total body water ↑ with
- ↓ age (neonates highest)
- Male gender
- ↑ volume of distribution for water soluble drugs.
- total body fat
- ↓’d in children and men → ↓ Vd of lipophillic drugs
- skeletal muscle bulk
- ↑’d in men → ↑ Vd
- disease
- Sepsis: alterations in pH alter ionised and bound fractions of drugs, increased permeability, organ failure and acute phase reactants all alter protein binding and Vd.
Measurement of Volume of Distribution
It is calculated based on the formula:
Since it is impossible to measure plasma concentration at time zero, an extrapolated plasma concentration is used.
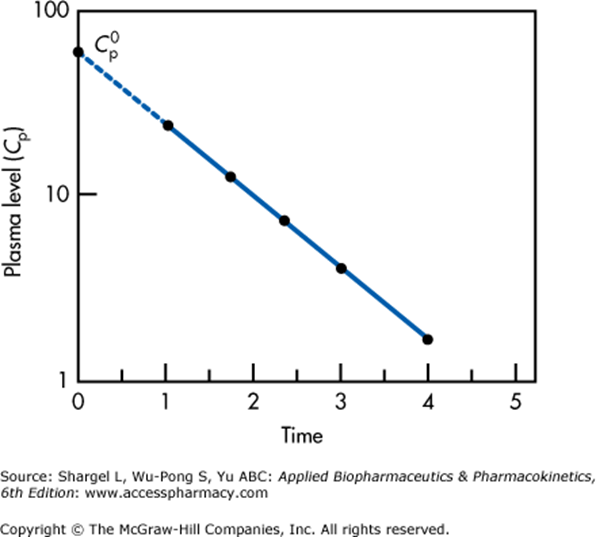
Semilogarithmic plot above illustrates extrapolation of plasma level to time 0 required to determine the volume of distribution
This graph applied for a single compartment model only.
For multiple compartments which will appear as a non-linear relationship, extrapolation back to t = 0 must be performed for each compartment separately.
Gladwin / JC 2019
Examiner Comments
2019A 05: 51% of candidates passed this question.
The first two parts of the question were reasonably done. Most candidates had well-structured answers which included drug factors and patient factors. In addition to listing the factors it was expected candidates state how these factors affect volume of distribution. Explaining how volume of distribution is determined was not so well done.
2015B 12: 35% of candidates passed this question.
A definition was required that included reference to plasma concentration, total body content / dose, and the theoretical nature of the volume which can exceed the physical volume of the body.
A broad answer listing patient and drug-related factors was required. Patient factors could include age, gender, muscle mass, fat mass and abnormal fluid distribution (oedema, ascites, pleural effusion). The drug factors would include tissue binding, plasma protein binding and physicochemical properties of drug (size, charge, pKa, lipid solubility, water solubility).
2018B 07
Describe protein binding and its significance in pharmacology.
CICMWrecks Answer
Proteins and drugs may be bound together by weak bonds. These include ionic bonds, van der Waal’s forces, and hydrogen bonds.
Protein Binding:
- Refers to the degree to which drugs attach to proteins in the body.
- most drugs which are protein bound, bind either albumin or a1-acid glycoprotein according to their pKa
- some bind specialized proteins e.g. steroid binding globulin, transcortin etc
- increases Vd and reduces free fraction of drug
- reduces renal clearance by filtration but not active secretion
- is a source of interactions
- E.g: Phenytoin 95%, Warfarin 97%
Drugs may bind to proteins in:
- Plasma
- Albumin: Binds acid and neutral drugs.
- High capacity
- Three major binding sites (six total)
- Site I: warfarin, bilirubin, salicylates, phenytoin, sulfonamides
- Site II: benzodiazepine, NSAIDs, penicillin
- digoxin, verapamil, quinidine
- level is reduced by
- catabolic states: burns, malignancy, renal/hepatic disease, pregnancy, old age, neonates
- α1-acid glycoprotein: Binds basic drugs (e.g. lidocaine, propranolol, TCA’s)
- Single binding site
- Low capacity
Typically results in lower total binding (compared to albumin) of alkaline drugs, despite its increased affinity. - level increased in catabolic states: burns, renal transplant, malignancy, trauma, inflammatory diseases: RA, UC, Crohn’s, myocardial infarct
- level decreased in pregnancy, neonates
- Lipoprotein E.g. Steroid binding globulin, transcortin
For lipid soluble drugs. - Alpha, beta and gamma globulins
- Albumin: Binds acid and neutral drugs.
- Tissue
- Receptor
Protein binding is important as:
- Only unbound drugs are able to:
- Cross cell membranes
- Interact with receptors
- Undergo metabolism
- Reduced protein binding increases clearance of drugs with low extraction ratios.
- Be filtered by the kidney
- Highly tissue bound drugs:
- Have a long duration of action
- Have a high volume of distribution, prolonging their elimination
- May build up in tissues, leading to adverse effects
e.g. Corneal deposition, lung fibrosis.
Protein binding is affected by:
- Affinity of drug for protein
- Ionised drugs do not bind to protein
pH. - Competition between drugs for binding sites
- Ionised drugs do not bind to protein
- Amount of protein
- Disease: Due to:
- Hypoalbuminaemia: Negative acute phase reactant.
- Increased α1-acid glycoprotein: Acute phase reactant.
- Competition: Source of pharmacokinetic interactions.
- Disease: Due to:
Protein binding typically:
- Correlates with lipid solubility
- Is important only when it is very high
- Results in a decreased VDss when plasma binding is high
- Results in an increased VDss when tissue binding is high
- Is important in duration of action as it also relates to affinity for tissue proteins
JC 2019
Examiner Comments
2018B 07: 19% of candidates passed this question.
Descriptions of protein binding were generally too brief (e.g. a statement saying that drugs and hormones bind to proteins in the plasma rather than a description of usually reversible binding with a drug-protein equilibrium).
It was expected that the factors which determine protein binding would be described. Marks were attributed if proteins, along with characteristics of the drugs they bind, were named. Candidates achieved better marks if they named the pharmacological parameters affected by protein binding and explained how and why change occurs along with the significance of those changes. Few candidates differentiated between tissue and plasma protein binding and the different effects on the volume of distribution
iv. Describe the mechanisms of drug metabolism and clearance.
2009B 17
Explain the difference and the clinical relevance, between zero and first order kinetics. (60% marks)
Give an example that is relevant to intensive care practice. (40% marks)
CICMWrecks Answer
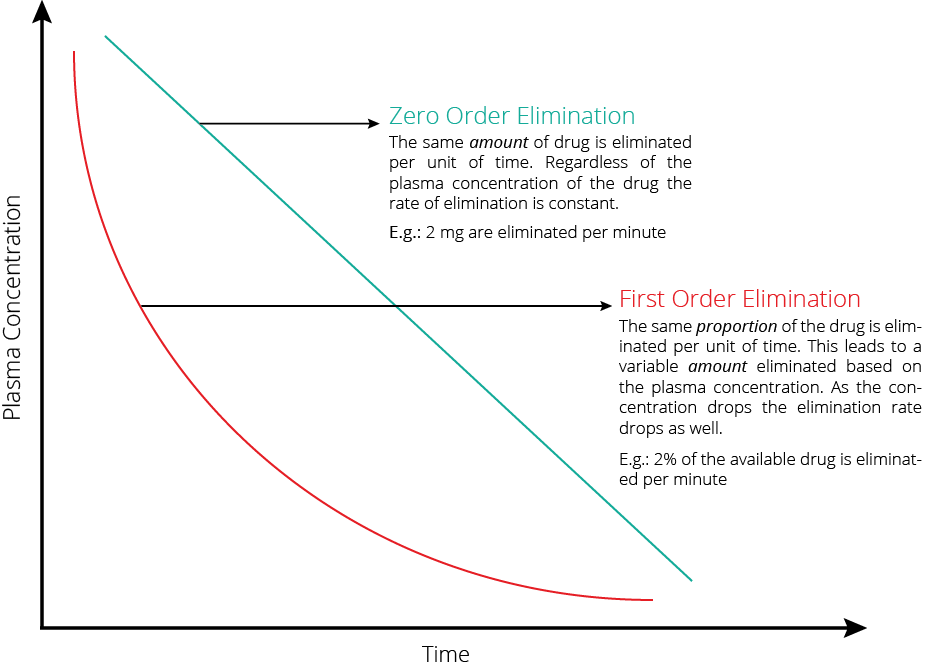
| Zero order kinetics | First order kinetics | |
|---|---|---|
| Definition | Rate of elimination = dP/dT = constant | dC/dt = kC |
| Elimination = constant | Rate of Elimination = Cl x C | |
| Explanation | • Elimination of a drug from the body is saturable. • The rate of change of concentration with time is constant (v) | • Rate of Elimination of a drug from the body is proportional to the concentration. • Elimination is equal to the area under the curve of the concentration versus time curve after dose of drug is administered. |
| Example | Phenytoin when plasma [ ] > 10 μg/mL Other examples include • Ethanol • Aspirin | Phenytoin when plasma [ ] > 10 μg/mL |
| Implication | • Small changes in doses or adjustment of other medications effecting pheytoin metabolism • Narrow therapeutic index, and zero order kinetics and therapeutic drug monitoring is important | Elimination is dependent on blood flow (ie high extraction ratio) |
Phenytoin or Aspirin
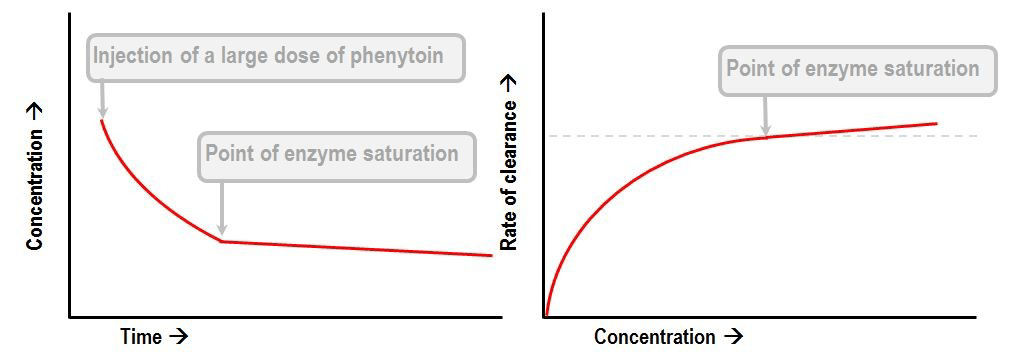
- Display Michaelis-Menten kinetics
- Zero order at high concentration when enzyme saturate
- First order at low concentrations
- Thus highly variable plasma concentrations
- Where
- S is the substrate concentraton
- Vmax is the maximum elimination capacity
- Km is the drug concentration at which the rate of elimination is half of the maximal rate.
Gladwin 2016
Examiner Comments
2009B 17: 2 (22%) of candidates passed this question
Candidates should expect that questions relating to “the pharmacology of ……” are likely to be common. Thus candidates should have prepared structured approach for any such question. For example, one that includes predefined major categories such as pharmacodynamics and pharmacokinetics and sub-categories such as mechanism of action, absorption, preparations, bioavailability, volume of distribution, metabolism, elimination, adverse effect, clinical indications, precautions/interactions, etc. and the information relevant to each category. Failure to take a structured approach to such questions, as was observed amongst some candidates within this exam, risks omission of vital facts (and not gaining marks) and errors. Noradrenaline is such a common drug within intensive care practice and so candidates would be expected to know it in great detail. There are many references for it, such as the ones listed below.
Syllabus – G3a, 2b
References – Goodman and Gillman Chp 10 and Katzung.
2019B 15
Define clearance and hepatic extraction ratio (30% of marks).
Describe the role of the liver in drug clearance with examples (70% of marks).
CICMWrecks Answer
Clearance (Cl)
Defined as the volume of plasma from which drug is completely removed per unit time (units – mL/min)
- Drug can be cleared from plasma via two routes:
- “Elimination” from the body – Drug excreted unchanged (via renal or biliary routes) and/or metabolised (by liver or other organ)
- Non-compartmental model: Cl = Dose / AUC
- Compartmental model: Cl=Vd x Kel = Vd x (ln2/t1/2)
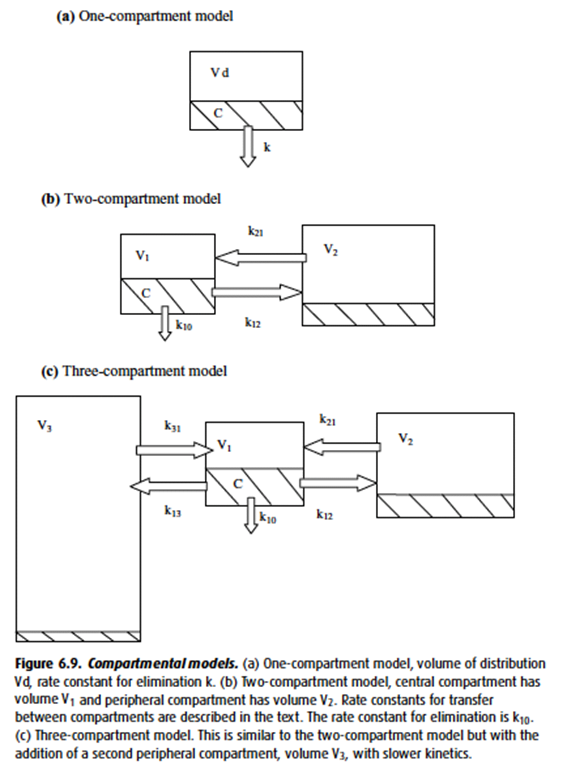
- “Intercompartmental clearance” (* occurs only in multi-compartment model *) – Drug distributes from the central to peripheral compartment(s) → determined by the rate constant for intercompartmental transfer (k12, k21; k13, k31; Etc.)
- “Elimination” from the body – Drug excreted unchanged (via renal or biliary routes) and/or metabolised (by liver or other organ)
- Significance of clearance:
- Determines maintenance dose rate needed to achieve a plasma [drug] at steady state
- “Maintenance dose rate” = Cl x desired [ ]PLASMA
Hepatic Extraction Ratio
Hepatic extraction ration (HER) – Fraction of drug that is irreversibly removed during 1st pass of blood through the liver → determined by intrinsic clearance (enzyme activity) and (ii) unbound % of drug
where
QH = Hepatic Blood Flow
ERHep = Hepatic Extraction Ratio
FU = fraction of drug unbound in plasma
ClInt = hepatic enzymatic capacity
Hepatic clearance
“Hepatic clearance” → determined by Hepatic Blood Flow and Hepatic Extraction Ratio
- Hepatic blood flow (HBF) – Rate at which drug is delivered to the liver
- Hepatic extraction ration (HER) – Fraction of drug that is irreversibly removed during 1st pass of blood through the liver → determined by intrinsic clearance (enzyme activity) and (ii) unbound % of drug
Hepatic clearance = HBF x HER = HBF x [(% unbound) x (intrinsic clearance)]
Important to note:
- Drugs with HER > 0.7 (↑ enzyme activity or “flow-limited”), such as GTN:
- Hepatic drug clearance is dependent on HBF (“perfusion-dependent elimination”)
→ ↑ HBF will lead to ↑ hepatic drug clearance - Changes in HER (intrinsic enzyme activity or unbound %) have minimal effect on hepatic drug clearance as heaps of drug is already removed at a given time
– i.e. Hepatic clearance ≈ HBF
- Hepatic drug clearance is dependent on HBF (“perfusion-dependent elimination”)
Other roles of liver in drug pharmacokinetics
Absorption (First pass metabolism):
- Drugs absorbed from GIT (except buccal and rectal mucosal) enter portal venous blood and pass through liver before entering systemic circulation
- They are metabolised by enzymes within the (i) liver (main) and (ii) gut wall (minor)
- FPM is a main reason why plasma [ ] after an oral dose is less cf. similar IV dose → as a result, it is a key determinant of oral bioavailability
- Significance – Drugs with ↓ FPM are either well-absorbed, stable in GIT, and/or have minimal hepatic metabolism → thus, have ↑ oral bioavailability (and ↑ plasma [ ]). The opposite is true for drugs with ↑ FPM
where,
FB = Bioavailable fraction
FA = Fraction absorbed
FG = Fraction remaining after gut mucosal metabolism
FH = Fraction remaining after hepatic metabolism
Metabolism:
Metabolism → process of chemically altering a drug within the body
Mainly occurs in liver (by hepatic microsomal enzymes), and few other sites
Effects of metabolism:
- ↓ drug activity (main effect): converts a “pharmacologically active” form of drug (Ie. non-polar and lipid soluble) into a “pharmacologically inactive” form (Ie. more polar and water-soluble) that can be excreted from the body (esp in bile or urine)
- ↑ drug activity: “Prodrug” → active moiety (Eg. enalapril → enalaprilat; parecoxib → valdecoxib)
- Produce metabolites with equal activity to parent compound (Eg. diazepam, propranolol)
Phases of metabolism:
- Phase I (functionalisation or non-synthetic):
- Alter drug reactivity for phase II reaction and to ↑ drug polarity/water-solubility
- Oxidation Including Hydroxylation (Eg. propofol), desulphation (Eg. STP), dealkylation (Eg. vecuronium), dehalogenation (Eg. volatiles), deamination, Reduction, Hydrolysis
- Phase II (conjugation or synthetic)
- ↑ water solubility of drug or its metabolite by conjugating it to a polar endogenous moiety (Eg. sulphate, glucuronyl, methyl, Etc.) → permits excretion in urine or bile
- Glucuronidation via glucuronosyltransferase (Eg. morphine, propofol)
- UDP-glucuronic acid is conjugated to the drug → conjugate is inactive and water-soluble → excreted in urine/bile
- Conjugated undergoes “enterohepatic recirculation” if eliminated in bile → intestinal bacterial glucuronidases hydrolyses glucuronide → liberates free drug which is reabsorbed back into circulation → results in prolonged drug action
- Other reactions: Sulphation, Acetylation, Methylation, Glutathione via glutathione-S-transferase (Eg. EtOH)
- Note – All these reactions involve non-microsomal enzymes, EXCEPT for glucuronidation (requires hepatic CYP450 microsomal enzymes)
Source: Bianca’s notes
JC 2019
Examiner Comments
2019B 15: 70% of candidates passed this question.
Clearance was generally well answered. It is the volume of plasma cleared of a drug per unit time, not the mass of drug cleared.
An equation was helpful in identifying the relevant components of hepatic clearance. ClHep=QH X ERHep ERHep= FU x ClInt / QH + FU x ClInt QH = hepatic blood flow ERHep = hepatic extraction ratio FU = fraction of drug unbound in plasma ClInt = hepatic enzymatic capacity
Many candidates did not describe the effects of hepatic blood flow and intrinsic clearance on drugs with high and low hepatic extraction ratios. Some discussion of Phase I and II reactions was also expected.
2016A 20
Outline the role of the liver in drug pharmacokinetics.
CICMWrecks Answer
This answer could be structured by splitting the content into major categories. These could be:
- the clearance functions of the liver
- first pass metabolism
- mechanisms of hepatic metabolism
- the effects of liver disease.
The role of the liver in drug clearance
- The role of the liver in pharmacokinetics is as an organ of clearance.
- The two major determinants of hepatic clearance are the efficiency of drug removal from the blood and the efficiency of blood delivery to the liver.
- Efficiency of drug removal by the liver is described by the hepatic extraction ratio, which is the fraction of the drug entering the liver in the blood which is irreversibly removed (extracted) during one pass of the blood through the liver.
- The hepatic extraction ratio is determined largely by the free (unbound) fraction of the drug and by the intrinsic clearance rate, which is the intrinsic ability of the liver to remove (metabolise) the drug in absence of restrictions imposed on drug delivery to the liver cell by blood flow or protein binding.
- The effect of liver blood flow on hepatic clearance depends on the hepatic extraction ratio of the drug.
- With increasing hepatic blood flow, hepatic extraction ratio will decrease for all drugs.
- For drugs with low intrinsic clearance:
- Hepatic extraction ratio will drop more rapidly with increasing hepatic blood flow
- Hepatic clearance will not increase significantly with increasing blood flow
- For drugs with high intrinsic clearance:
- Hepatic clearance will increase in a fairly linear fashion, in proportion to hepatic blood flow
- Increasing the intrinsic clearance will have diminishing effect on total hepatic clearance
The effect of the liver on first pass metabolism
- First pass clearance is not just hepatic but is a combination of metabolism by gut bacteria, metabolism by intestinal brush border enzymes, metabolism in the portal blood and metabolism by liver enzymes.
- For drugs with low hepatic extraction ratio:
- First pass clearance will be low
- Changes in hepatic enzyme function will have little effect on first pass clearance
- For drugs with high hepatic extraction ratio:
- First pass clearance will be high
- Changes in hepatic enzyme function will have a significant effect on first pass clearance
Biotransformation in the liver
By convention, the metabolic functions of the liver are divided into Phase I and Phase II reactions
- Phase I reactions:
- Examples of Phase I reactions:
- Hydrolysis
- Reduction
- Oxidation.
- Characteristics of Phase I reactions:
- these reactions expose or introduce a functional group (–OH, –NH2, – SH or –COOH)
- They usually result in a small increase in hydrophilicity.
- Examples of Phase I reactions:
- Phase II reactions:
- Examples of Phase II reactions:
- Glucuronidation
- Sulfation
- Acetylation
- Methylation
- Conjugation with glutathione
- Conjugation with amino acids eg. taurine, glutamine, glycine
- Characteristics of Phase II reactions:
- The products are supposed to be significantly more hydrophilic than the original substrate
- Examples of Phase II reactions:
Effects of changes in liver function
- The effects of changes in synthetic function
- The liver synthesises plasma proteins; plasma protein binding influences the volume of distribution
- Low plasma protein levels lead to raised free drug levels (the free fraction increases)
- This process is therefore synergistic with the concurrent decrease in liver blood flow and hepatic extraction ratio
- The liver synthesises plasma esterases and peptidases; these metabolise certain drugs
- Significant liver disease can result in prolonged clearance of drugs which are susceptible to these enzymes (eg. suxamethonium)
- The effect of changes in secretory function
- Drugs and metabolites which rely on biliary excretion will be retained, and may require dose adjustment
- Drugs which enjoy enterohepatic recirculation may have decreased halflives due to failure of recirculation
- High bilirubin levels may result in the displacement of drugs from albumin as it competes for binding sites
- Decreased secretion of bile may result in malabsorption
- The effects of portal hypertension on pharmacokinetics
- Portal venous hypertension leads to shunting of portal venous blood into the systemic circulation
- This has the effect of decreasing first pass metabolism
JC 2019
Examiner Comments
2016A 20: 62% of candidates passed this question.
Most candidates structured their answer to this question well – they were aware of first pass metabolism and the effect of protein synthesis upon volume of distribution of drugs. Knowledge concerning Phase I and Phase II reactions was frequently inadequate. Many candidates were aware that these processes as well as inactivating or activating drugs resulted in increased water solubility to aid excretion via bile or urine. Few candidates discussed the significance of the large blood flow to the liver or the implications of high and low extraction ratios especially in relation to liver blood flow.
2012B 05
How does liver failure affect the pharmacology of drugs?
CICMWrecks Answer
Pharmacodynamics
- Altered receptor binding
- Postulated for relative resistance to non-depolarizing NMB in liver failure
Pharmacokinetics
- Absorption
- Portal hypertension and interstitial oedema reduce absorption of drugs
- Hepatic dysfunction
- Increased bioavailability of drugs susceptible to hepatic first pass metabolism (diazepam)
- Decreased activation of pro-drug (clopidogrel requires CYP2C19 activation)
- Distribution
- Decreased synthesis of plasma proteins
- Increases free fraction of protein bound drugs
→ Unbound drug more permeable → increased volume of distribution (amiodarone)
→ Unbound drug more active
- Increases free fraction of protein bound drugs
- Hypervolaemia secondary to renal Na+ and H2O retention
- Increased Vd for drugs distributed to extracellular volume
- Decreased pH alteres ionization of acids and bases (acids ionized when pH above pKa and bases ionized when pH below pKa)
- Decreased synthesis of plasma proteins
- Metabolism
- Decreased hepatic metabolism
- Increases T1/2b of hepatically metabolized drug → Accumulation of drug if no alteration
- Decreased butylcholinesterase production → Decreased metabolism for drugs metabolised by plasma esterases (suxamethonium – however clinically not significant for this drug) – red cell esterases remain therefore no effect for remifentanyl
- Decreased hepatic metabolism
- Elimination
- Hepatorenal syndrome → decreased renal clearance
- However renal drug clearance tends to decrease even in absence of renal impariment (drugs not hepatically converted to more water soluble metabolites)
- Biliary clearance of drug is reduced
Sakurai 2016
Examiner Comments
2012B 05: 13 (59.1%) of candidates passed.
Good answers were structured using pharmacokinetic and pharmacodynamics headings. They included some mention of changes in absorption, volume of distribution (an increase in Vd in liver failure), altered protein binding, altered metabolism and thus change in clearance, and changes in excretion (decreased biliary excretion of drugs). In respect to pharmacodynamics candidates could have mentioned increased sensitivity and prolonged action of sedative drugs, oral anticoagulants, etc. Good candidates also differentiated for acute (often hepatocellular dysfunction) and chronic liver failure (cirrhosis and changes in liver blood flow). Common problems were not using a logical structure to answer the question and stating an effect but not describing how this affected pharmacology. For example stating decreased albumin production but then not stating the consequence of this on drug distribution. Primary examination questions may often require candidates to integrate knowledge from across different sections of the syllabusor apply basic physiological or pharmacological principles.
2016A 08
Describe the characteristics of a drug that influence its excretion by the kidneys.
CICMWrecks Answer
Excretion by kidneys can be influenced by drug characteristics
1) Which affect filtration at glomerulus
2) Which affect secretion into tubules
3) Which affect reabsorption in the tubules
1) Which affect filtration at glomerulus
Filtration at the glomerulus – Dependent on Filtration fraction [GFR / RBF] and net starling force.
Normal filtration is 180 L/day (20% of renal plasma flow)
- Protein binding:
- Only free drug present in filtered plasma will be excreted.
- Concentration of filtered drug will be the same as in unfiltered plasma
- Highly protein bound drugs are poorly filtered
There is only a weak concentration gradient favouring dissociation from plasma proteins.
- Molecule size:
- Size:
- Molecules larger than 30 Angstrom is not filtered
- Weight:
- Substances less than 7,000 Da are freely filtered
- Substances greater than 70,000 Da are essentially impermeable
- Size:
- Hydrophilic/lipophobic
Lipophilic drugs may be filtered at the glomerulus but will be freely reabsorbed during their passage down the tubule, such that only trivial amounts are eliminated in urine. - Charge:
- Positive ions are filtered (Glomerulus is slightly negative charged)
2) Which affect secretion into tubules
Active process, allows secretion against concentration gradients
- Acid/Base:
- Saturable process – Saturation may occur of a basic transporter whilst still allowing excretion of acidic drugs, and vice versa.
- Size:
- Molecules which are too large to be filtered in the glomerulus may still be cleared renally by these mechanisms
- Protein binding:
- Protein-bound drugs are not cleared this way; only the unbound fraction is available for active secretion.
3) Which affect reabsorption in the tubules
Passive diffusion down a concentration gradient.
- Hydrophilic molecules can only be reabsorbed by a specialised transport mechanism
- Drug ionization:
- Only non-ionised drug can be reabsorbed passively
- Acidic drugs will become ionised in an alkaline urine (and vice versa), reducing their solubility
This is the physiological justification for urinary alkalinisation.
JC 2019
Examiner Comments
2016A 08: 29% of candidates passed this question.
Drug characteristics that might influence the renal excretion processes include charge, size, solubility, and binding to specific structures or protein. Whether the drug is unchanged versus metabolised can influence these factors.
This question tests core knowledge of pharmacology principles and should be answered with equations, graphs or simple clear descriptions of physical and chemical principles. Extended examples and hedged statements about “influencing” without the direction, magnitude and necessary conditions for the influence did not score marks
2011B 03 – 2010A 08
Describe the role of the kidney in drug excretion and the factors affecting this (80% marks).
Briefly outline how you would alter the dosing of a drug with high renal excretion in a patient with renal impairment (20% marks)
2008A 17
Describe the role of the kidney in drug excretion, and the factors affecting this.
Briefly outline how you would alter the dosing of gentamicin in a patient with renal impairment.
CICMWrecks Answer
Renal Drug Excretion
Due to glomerular filtration and balance of secretion and reabsorption
Glomerular Filtration
- Physiological factors
- Starling forces
- Filtration pressure ∝ (PG – PBC) – ( πG – πBC)
Where- PG = Glomerular hydrostatic pressure (60mmHg)
- PBC = Bowman’s Capsule hydrostatic pressure (18mmHg)
- πG = Glomerular oncotic pressure (32mmHg)
- πBC = Bowman’s Capsule oncotic pressure (Negligible)
- Filtration decreases
- Shock → decreased glomerular pressure
- Obstruction → increased bowman’s capsule hydrostatic pressure
- Hypoproteinaemia → hepatic failure, nephrotic syndrome
- Fick’s Law (see eqn)
- Filtration increases with decreased MW, increased concentration gradient
- Filtration decreases with increased GBM thickness (glomerulosclerosis, deposition) and loss of glomerular surface area (usually 0.8m2. may be lost with renal failure)
Fick’s Law:
- Drug factors
- Size (Due to filtration slits and podocyte foot processes)
- Particles <4nm freely filtered
- Particles >8nm excluded
- Charge
- GBM has negative charge, therefore negatively charged molecules repulsed → decreased filtration
- Protein binding
- Highly protein bound drugs inhibited from filtration due to large size and charge of proteins
- Size (Due to filtration slits and podocyte foot processes)
Secretion
- Due to specific transporters
- P-glycoprotein (Amphipathic anions)
- MPR-2 (Conjugated metabolites)
- ATP-Binding Cassette Transporters (Organic cations)
Reabsorption
- Passive reabsorption occurs in PCT and DCT
- Membrane transporters for facilitated transport are present in DCT
- Ionization
- pH of urine may lead to drug “trapping”
- Acids ionized with pH > pKa
- Bases ionized when pH < pKa
- pH of urine may lead to drug “trapping”
- Lipophilicity
- Allows permeation through phospholipid bilayers of cell membrane → reabsorption
Dose adjustment of renally excreted drugs
- According to clearance and GFR
- Loading dose = Target concentration x Vd
- Usually no alteration of loading dose required
- In ESRF, volume of distribution may be increased → loading dose may need to be increased
- Dosing rate = Target concentration x Cl
- As clearance is reduced in renal failure dosing rate requires reduction
- Maintenance dose = Dosing rate x Dosing interval
- Maintenance dose and/or the dosing interval may need to be reduced (depending on whether AUC or peak concentration of drug is required for drug effect)
- Plasma monitoring of [drug] concentration required for drugs with low therapeutic index
Dose adjustment of Gentamicin
MoA: Inhibition of bacterial protein synthesis through irreversible binding to the 30s bacterial ribosome
Has a narrow therapeutic window, hence dose optimization and therapeutic drug monitoring are crucial.
| Concentration dependant killing | Active concentration needs to be acheived (~8-10 times Minimim Inhibitory Concentration/MIC) Time above MIC does not need to be maintained | Loading dose should not be altered even in renal failure (5-7mg/kg) |
| Significant postantibiotic effect Distribution: Hydrophilic, so Distribute mainly to extra-cellular fluid. Redistribution can occurs upto 16-24 hours. | Once daily dosing sufficient without renal failure | |
| ESRF – Volume of distribution theoretically larger (but in case of Aminoglycosides, might be lower – suspected due to tissue displacement by other molecules like urea) | Loading dose adjustment in ESRF 4mg/kg | |
| Metabolism: Nil Excretion: Rapidly excreted by glomerular filtration. Hence accumulate in renal failure (t1/2 upto 30-60hrs) | Subsequent Dosing interval based on renal clearance (GFR / CrCl) Frequent dosing only increases toxicity with no improvement in efficacy. | Maintenance dosing less frequent in renal failure Dose interval based on pre-existing guidelines, and with monitoring of pre-administration levels CrCl 40 to 59 mL/minute: Administer every 36 hours CrCl 20 to 39 mL/minute: Administer every 48 hours CrCl <20 mL/minute: Monitor serum levels and redose when gentamicin level is less than 1 mg/L |
| End stage renal disease / Dialysis (t1/2 upto 100hrs) | Check level prior to dialysis, and only administer dose after dialysis (based on expert advice) | |
| Low Therapeutic index | Frequent monitoring of levels required | |
Sakurai / JC 2019
Examiner Comments
2011B 03: 13 (52%) of candidates passed this question.
It was expected candidates would expand on the role of the kidney in the excretion of drugs and metabolites. A statement that referred to the classical features of drugs that undergo renal excretion (eg polarity, lipid solubility, size and protein binding, drug metabolites) and a definition of renal clearance was expected and would have provided an excellent introduction to any answer. It was then expected that candidates would mention, in some detail, the processes of filtration and secretion (both active and passive and both proximal and distal along the renal tubules). The question also asked for factors that affect renal drug excretion, for example, a reduction in GFR or alteration in protein binding. An approach to alterations of dosing would require some consideration of assessing degree of dysfunction (GFR estimation / calculation) then an understanding that it would not impact on loading doses but would influence subsequent dosing of renal cleared drugs. Plasma monitoring provides useful information for some drugs, particularly those with a narrow therapeutic index.
Syllabus: II2d, D12h
Recommended sources: Goodman and Gilman’s the Pharmacological Basis of Therapeutics pgs10-14; Foundations of Anaesthesia Basic and Clinical Science, Hemmings pg107; Basic and Clinical Pharmacology, Katzung pgs 35, 48-49.
2010A 08: 0 (0%) of candidates passed this question
The preponderance of marks was allocated to a discussion of renal drug excretion and factors altering this function. Detail was expected including a definition of clearance, and the balance between filtration / secretion / reabsorption in the tubules. Specific mention of GFR, molecular weight of filterable compounds, protein binding, and charge effects determining filtration at the glomerular level was anticipated. Tubular transport mechanisms for secretion and reabsorption in the proximal and distal tubule should have been included in the discussion.
Candidates needed to cover factors which alter GFR, competition for transport proteins, changes in pH on drug elimination, and disease states in answering the question.
An understanding that drug dosing should be based on estimating creatinine clearance and plasma concentration monitoring was essential. Loading dose is usually unaltered. However, maintenance dose and dosing interval need to be adjusted owing to an increased half-life. Many candidates did not emphasise the need to increase dosing interval as well as reduce maintenance dose in renal impairment.
Syllabus: II2d, D12h
References: Goodman and Gilman’s the Pharmacological Basis of Therapeutics p10-14. Foundations of Anaesthesia Basic and Clinical Science, Hemmings p107.
Basic and Clinical Pharmacology, Katzung p35, 48-49.
2008A 17: No candidates (0%) passed this question.
The main points candidates were expected to cover included a brief definition of renal clearance followed by a description of the drug factors that affect this (filtration, secretion and reabsorption), recognition that GFR and protein binding was important in the answer. A brief description of gentamicin kinetics that affect dosing regimens and a statement that dosing would be guided by calculated GFR and measured drug levels wold round out a good answer. Correct elaboration of the above factors were rewarded with additional marks.
Candidates failing this question submitted answers where concepts were randomly mentioned with no attempt to integrate these into a cohesive answer that demonstrated an understanding of the topic. Writing random words without examples or explanations did not demonstrate sufficient understanding to be rewarded with marks. Again, many answers lacked sufficient detail in the answer.
Syllabus – Pharmacokinetics
v. Explain the kinetics of an intravenous bolus and infusion.
2009A 10
Describe the calculations involved in determining the loading dose and maintenance dose for an intravenous infusion (50% of marks). What factors may affect these values in the critically ill (50% of marks)?
CICMWrecks Answer
Continuous IV Infusion
- Volume of distribution
- Apparent volume into which a drug disperses in order to produce the observed plasma conc
- The physicochemical properties of a drug influence Vd
- lipid solubility – highly lipid soluble drugs have larger Vd
- charge characteristics – highly charged drugs have smaller volumes of distribution
- tissue binding may result in increased Vd
- pathology – renal and hepatic disease leads to increased Vd due to fluid changes
- Plasma concentration
- is the amount of drug/volume within which it is diluted
- Clearance
- is the volume of plasma from which the drug is cleared from per unit time (usually ml/min)
- it is also the dose/area under the curve
- Loading dose (mg)
- aims to achieve a required plasma concentration
- therefore = desired peak concentration (mg/L) × clearance (L/hr)
- (divided by bioavailability if not IV)
- Maintenance dose (mg/hr)
- aims to achieve a constant plasma concentration range
- is equal to the elimination of the drug
- Is desired peak concentration (mg/L) × clearance (L/hr)
Factors which affect loading dose and maintenance dose in critically ill
| Effects of critical illness | Impact on loading dose and maintenance dose |
| VOLUME OF DISTRIBUTION | |
| Increased Vd (due to fluid overload) | Increased loading dose and maintenance dose or dose rate |
| Decreased Vd (due to hypovolaemia) | Decreased loading dose and maintenance dose or dose rate |
| CLEARANCE | |
| Decreased renal clearance (due to decreased renal blood flow or renal parenchymal damage) | Decreased maintenance dose or dose rate; also possibly increased dosing interval. Loading dose could remain unchanged |
| Increased renal clearance (hyperdynamic states, eg. early sepsis) | Increased maintenance dose or dose rate; also possibly dencreased dosing interval Loading dose could remain unchanged |
| Decreased hepatic clearance (decreased hepatic blood flow or inhibited liver enzyme function) | Decreased maintenance dose or dose rate; also possibly increased dosing interval. IV loading dose could remain unchanged Oral loading dose would need to be decreased to accommodate for the decreased first pass metabolism |
| Increased hepatic clearance (increased hepatic blood flow or hepatic parenchymal clearance) | Increased maintenance dose or dose rate; also possibly decreased dosing interval. IV loading dose could remain unchanged Oral loading dose would need to be increased to accommodate for the increased first pass metabolism |
| BIOAVAILABILITY | |
| Decreased protein binding (due to lower levels of protein) | Increased free unbound fraction of the drug, which gives rise to increased clearance and increased drug effect. |
| Decreased gut absorption (due to decreased splanchnic blood flow and/or decreased peristalsis) | Variable and inconsistent absorption of an otherwise correctly calculated oral loading dose |
| Competition for protein binding (eg. where bilirubin competes for albumin binding sites) | Increased free unbound fraction of the drug |
Examples of drugs illustrating an understanding of pharmacokinetics
- Theophylline: a drug which is dosed every half-life (300mg every 8 hours), which is equivalent to a dose rate of 37.5mg/hr, which is in turn equivalent to an infusion rate of 37.5mg/hr.
- Phenobarbitone: a drug with a vast volume of distribution, where the loading dose would be massive and toxic
- Morphine: a drug which is poorly orally bioavailable; an example of how the oral loading dose is affected by bioavailability
- Phenytoin: a drug which is highly protein-bound, and which is highly affected by the low plasma albumin associated with critical illness (thus, with low total drug levels the levels of free unbound drug may still be therapeutic)
- Corrected Phenytoin = Measured Phenytoin Level / ( (adjustment x albumin) + 0.1)
- Adjustment = 0.2; In patients with Creatinine Clearance < 20, adjustment = 0.1.
- Gentamicin: a drug which is cleared rapidly by the kidneys, a clearance which is significantly affected by poor renal function. It is an example of how the loading or maintenance dose should remain unchanged; instead the dosing interval should be extended.
- Vancomycin and β-lactams: examples of drugs which are subject to increased renal clearance in the context of hyperdynamic circulatory states, for example in early sepsis.
Source: Deranged Physiology
JC 2019
Examiner Comments
2009A 10: Pass rate: 30%
Main points for a pass included the equations for determining the loading and maintenance doses. Points were awarded for explaining the rationale for giving a loading dose and for relevant diagrams.
Answers to the second part of the question often lacked detail. Candidates should have mentioned alterations in volume of distribution, plasma proteins, renal & hepatic function. Examples of drugs illustrating an understanding of pharmacokinetics attracted extra marks.
Syllabus II 2 f
Reference: Rang Ritter Dale p120-123
vi. Describe the concepts of effect-site concentration and context sensitive half-time.
2012B 11
Discuss the pharmacokinetic factors that affect drug half-life.
CICMWrecks Answer
Definitions
Half life: Time taken for a reduction in one half the total ammount of drug from the body.
Half time: Time taken for a reduction in one half the plasma concentration of a drug
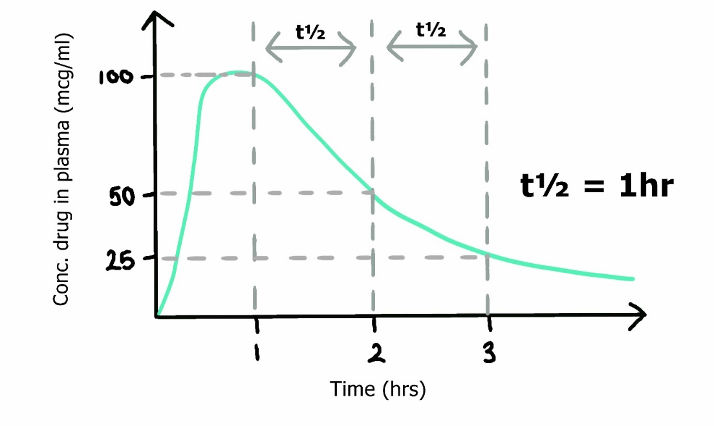
Simplified equation for half life
- Where
- Vd is volume of distribution of a drug
- Cl is its clearance
- ln(2), the natural log of 2, can be approximated as 0.693
Pharmacokinetic factors
Absorption
- Minimal impact on T1/2
- If initial plasma concentration saturates its metabolic pathway, clearance will become zero-order and rate of elimination will be independent of drug concentration
Distribution
- In the multi-compartment pharmacokinetic model,
- Drug initially entering the central (plasma) compartment will redistribute to other compartments (tissues), decreasing the amount remaining in the central compartment.
- As Vd increases, the ammount redistributing to other tissues increases → increased distribution away from central compartment → T1/2α (Distribution half time) decreases.
- This reduction in plasma concentration will decrease the concentration of drug reaching its metabolic organs (unless plasma metabolism) and prolong the elimination half-life.
- Drugs with high volumes of distribution display context-sensitive half time
- Protein binding of drug increases half life
- Prevents glomerular filtration
Metabolism and elimination
- Clearance is the volume of plasma completely cleared of drug per unit time
- Total body clearance is a sum of renal clearance (via metabolism or elimination), hepatic clearance (via metabolism, biliary secretion) and other mechanisms (such as lung metabolism, plasma cholinesterases etc.)
- As drug metabolism and elimination increases, T1/2β decreases
Zero-order kinetics
- As elimination of drug is independent of drug concentration, half life is not constant and progressively shorten by half
- E.g. 100mg of drug in body eliminated at 10mg/hr hour
- Half life from 100mg (to 50mg) would be 5 hours
- Subsequent half life (from 50mg to 25mg) would be 2.5 hours etc.
- E.g. 100mg of drug in body eliminated at 10mg/hr hour
Sakurai 2016
Examiner Comments
2012B 11: 10 (45.4%) of candidates passed.
Half-life (t½) is the time required to change the amount of a drug in the body by one-half during elimination. Candidates were expected to discuss the two main factors which affect drug half-life, namely volume of distribution and clearance. Marks were awarded for the formula (t½ = 0.693 x Vd /CL), the factors which affect the volume of distribution and drug clearance but not for a discussion of factors affecting drug absorption.
vii. Explain clinical drug monitoring with regard to peak and trough concentrations, minimum therapeutic concentration and toxicity.
2008A 04
Describe the factors that are important when interpreting plasma drug concentrations.
CICMWrecks Answer
FACTORS
Factors may be pharmacokinetic, Pharmacodynamic or Patient factors
1) Pharmacokinetic factors
- Absorption:
- enables an appreciation of bioavailability and variation in different routes
- heparin is an example, there is reduced absorption SC due to endothelial protein binding
- Distribution :
- highly lipid soluble drugs generally have a high volume of distribution
- plasma levels will therefore appear markedly decreased when compared to dose
- Protein Binding:
- Total plasma levels may not be reflective of active drug level
- E.g: Phenytoin and hypoalbuminemia (Low total, but enough free unbound drug)
- the pKa may be relevant as the amount ionised may be more important in determining
- effect rather than the total plasma level (ability to cross membranes)
- Effect site concentration may not be adequate inspite of plasma concentration
- (E.g Vancomycin in Ventriculitis, where high plasma levels are no guarantee of bactericidal CSF levels)
- highly lipid soluble drugs generally have a high volume of distribution
- Metabolism:
- drugs may have saturable metabolic pathways such that even at high doses the drug will
- be cleared at a fixed rate (phenytoin)
- co-administration of drugs may upregulate metabolic catalysts (e.g. CYP450s) increasing
- metabolism, or compete for substrate/catalyst decreasing metabolism
- Excretion:
- drugs that are excreted renally may have much higher plasma concentration if there is impairment or oliguria.
- Measurement related factors:
- Measurement Assay used
- Timing of sample collection
- In relation to dosing: Peak, trough, etc
- In relation to steady state concentration: Caution when interpreting before 3-5 half lives
2) Pharmacodynamic factors
- Relationship of plasma concentration to clinical drug effect:
- E.g. There is no relationship in the case of Levetiracetam
- Active Metabolites
- Plasma concentration may not correlate to clinical effect in the presence of Active metabolites
- Drug action receptor sensitivity
- action at the sensor (e.g. competitive antagonist vs non-competitive antagonist)
- Appreciation of reference values for plasma concentration levels
- To check therapeutic effect
- antimicrobials -minimal inhibitory concentration levels
- To monitor side effects
- local anaesthetic agents -CNS:CVS ratios
- To check therapeutic effect
3) Patient factors
- Age (decreased sedative concentrations required in elderly)
- Lean body mass vs toal body mass (lipid soluble versus small VD drugs)
- Renal and hepatic function (may prolong and elevate plasma concentrations)
- Pharmacogenetic factors
- abnormalities in CYP450 (e.g. variable codeine metabolism)
- abnormal plasma esterases – suxamethonium metabolism
- Individual variability in Drug Response
CLINICAL RELEVANCE
- Drug plasma concentrations are relevant when certain criteria are met:
- demonstrated relationship between drug concentration and therapeutic or toxic effect (digoxin)
- narrow therapeutic window (carbamazepine)
- variability in plasma level such that response cannot be predicted from dose alone (warfarin)
- drug produces effects, intended or unwanted, that are difficult to monitor (heparin)
- Convenient to collect, Fast and Accurate results
- Good benefit to cost
JC 2019
Examiner Comments
No candidates (0%) passed this question.
- The majority of the information required for this question is covered within the general pharmacology section of the syllabus. The main points expected for a pass were:
- Mention and discussion of pharmacokinetic factors such as drug absorption, volume of distribution, clearance, protein binding, dosing frequency and drug level sampling.
- Mention and discussion of pharmacodynamic factors such as drug sensitivity, and therapeutic range.
- Clinical relevance of a drug concentration (e.g. peak or trough level, total or free drug, etc).
- Candidates often failed to frame their answer to the question that was asked. Candidates could have made a greater use of illustrations and examples of drugs to help answer the question.
- Among other relevant listed references, candidates should seek information from within the text books – Basic and Clinical Pharmacology by B. G Katzung and Pharmacology by H. P Rang, J. M Ritter and M. M Dale.
viii. Describe the pharmacokinetics of drugs in the epidural and subarachnoid space.
2009A 12
Outline the pharmacology of an opioid injected into the spinal intrathecal space.
CICMWrecks Answer
Pharmacokinetic Effects
- D: Dependent on degree of lipid solubility. Fentanyl has > systemic absorption than Morphine. More likely to cause systemic opoid effects
- M: Minimal metabolic effects occur in the intrathecal space
- E: Clearance from the intrathecal space is via the arachnoid villi.
Pharmacodynamic Effects and Side-effects
Secondary to:
- Local Effects
- Analgesia
- MOP in spinal cord → ↓pain transmission primary afferent neurons
- Orthostatic hypotension
- 2° direct SNS blockade at sympathetic chain → vasodilation → venous pooling
- ↓ temperature
- Inhibition of shivering
- Analgesia
- Cephalic migration:
- Depends primarily on the lipid solubility of the drug
- Sedation
- Direct effect on μ-receptors in reticular formation
- Confusion
- Anticholinergic (pre-synaptic blockade of ACh release)
- Pruritis
- Interaction with μ-receptors in trigeminal nucleus
- CNS Excitation
- In overdose → myoclonic jerks
- Due to bulk flow to brainstem/basal ganglia
- ↓MV
- Direct opioid effect depressing ventilatory centre of brainstem
- ↑PaCO2 due to right-shift in ventilation v PaCO2 curve
- N&V
- Systemic Absorption
- ↓gastric emptying, ↓gut motility
- ↓MAP
- Mast cell degranulation → histamine release → vasodilation (rare with fentanyl, ↑with morphine)
Gladwin 2016
Examiner Comments
2009A 12: Pass rate: 30%
Though it would be unusual for patients to receive spinal opioids whilst they are in intensive care, the complications of spinal opioids are not an uncommon reason for admission to intensive care thus it is important candidates understand their pharmacology. Answers generally lacked structure. Outlining pharmacology should include pharmacokinetics, pharmacodynamics and side effects (both common and dangerous). An explanation of the effect of lipid solubility was expected. Following a structure will ensure a more complete answer.
Syllabus G2d2e
Reference: Neural Blockade. 3rd edition Cousins and Bridenbaugh
VIVAs
| 2014B | Dose response curves, renal clearance |
| 2012A | Thiopentone, conc vs time curves, Vd and factors, elimination, half life |
| 2011A | Oral drug absorption, basic pharm |
| 2010B | Basic pharm, opioid drugs |
| 2010A | Dose response curve and basic pharm including ED50, Com & non Comp antagonists |
| 2009A | pharmacokinetics in: Alcohol, Ageing |
Recent Comments